網羅的なゲノム解析から新たながんの原因遺伝子が明らかに
遺伝子変異のパターン分析からがんの原因解明や予防につながる国際共同研究成果が国際科学誌Nature電子版8月15日号に掲載
2013年8月15日
独立行政法人国立がん研究センター
本研究成果のポイント
- 国際的ながんゲノムプロジェクトのもと、約7,000症例のがんゲノムを網羅的に解析することでがんの原因となる体細胞変異のパターンを20種類以上発見
- 新たな発がんの要因となる遺伝子異常としてAPOBEC遺伝子群を同定
- 既知の発がん要因や疫学データ、臨床データなどを活用するがんの解明とがん予防に向けた新たな研究手法の確立に期待
独立行政法人国立がん研究センター(理事長:堀田知光、東京都中央区)は、国際的ながんゲノム研究共同プロジェクトにおいて、30種類のがん(7,042症例)からゲノムデータを収集し、体細胞突然変異のパターンを解析し、新たな遺伝子変異のパターンを明らかにするとともに、発がんの要因となる遺伝子異常を発見しました。本研究は、国立がん研究センター研究所(中釜斉所長)がんゲノミクス研究分野の柴田龍弘分野長を中心とする研究チームが、国際がんゲノムコンソーシアム(ICGC)(注1)のプロジェクトの一環として進めました。(本研究は、独立行政法人医薬基盤研究所先駆的医薬品・医療機器研究発掘支援事業および独立行政法人国立がん研究センターがん研究開発費による支援によって行われました。)
がんは遺伝子の病気であり、正常の遺伝子(DNA)に傷(突然変異)が蓄積した結果として発症することがわかっています。最新の高速シークエンサー(次世代型シークエンサー)(注2)技術を用いて、世界的な大規模がんゲノム解読プロジェクトのもと、膨大ながんゲノムビッグデータを解析し、さまざまながん種における突然変異パターンを分析することで、それぞれのがんにおける発がん要因を研究することが可能になってきています。
今回の国際共同研究(日本、英国、米国、ドイツ、フランス、スペイン、オーストラリアの7ヵ国が参加)において、30種類のがんから7,042例のがんゲノムデータを収集し、総計約500万個(4,938,362個)の体細胞突然変異を用いて、各がん種においてどのような体細胞突然変異パターンの組み合わせがどの程度貢献しているか、について解析を行いました。その結果、1.がんにおける体細胞変異のパターン抽出から、全部で22種類のパターンを発見しました。また、2.既知の発がん要因に加え、新たな要因としてAPOBEC遺伝子群(注3)の異常によるものが認められました。
今回の研究結果から、発がん要因の異なるさまざまながんにおける突然変異パターンの分類が可能になりました。今後発がん物質への暴露に関する疫学データや発がんモデル動物を活用することによって、それぞれのがんにおける発がん要因を推定していくことが可能になると考えられます。従って本解析結果は、発がんの分子機構の解明のみならず科学的エビデンスに基づくがん予防のための基礎的な知識基盤となることが期待されます。また当センターで進められている、がん患者の臨床検体の遺伝子プロファイリング研究においても、こうした解析手法の活用が進められることが期待されます。
本研究成果は、英国の科学雑誌『Nature 』の掲載に先立ち、オンライン版(外部サイトにリンクします)(8月14日英国時間18時掲載:日本時間8月15日午前2時)に掲載されます。
背景
がんは遺伝子の病気であり、正常の遺伝子(DNA)に傷(突然変異)が蓄積した結果として発がんが起こります。がん細胞で起こるDNA変異(体細胞突然変異)は、細胞分裂のときのDNA複製の誤り、外因性あるいは内因性発がん物質の暴露、DNAの酵素的修飾、DNA修復系の異常といったものが原因として起こることが知られています。一部のがんでは、発がん物質の暴露(喫煙による肺がんや紫外線による皮膚がん)、あるいはDNA修復系の異常(ある種の大腸がん)が体細胞突然変異の主要な原因であることが明らかになっていますが、多くのがんでは体細胞突然変異の発生とその蓄積過程について詳細は不明です。
個々の発がん要因(発がん物質)やその分子機構によって、結果として引き起こされる体細胞突然変異の組み合わせ(4種類の塩基[アデニン:A、チミン:T、シトシン:C、グアニン:G]がそれぞれ異なる3種類の塩基に変化する合計12種類)の組み合わせには特徴があることが知られています。例えば、たばこに含まれる発がん物質によって引き起こされる体細胞突然変異はCがAに(C>Aと表記)、あるいはGがT(G>T)に変化する2種類が大部分を占めています。
DNA解読技術の飛躍的な進歩に伴い、最新の高速シークエンサー(次世代型シークエンサー)によって、がんゲノムを1日で全解読することが可能になり、ゲノム全体あるいはたんぱく質をコードしている領域(エクソン領域)全体を丸ごと解析し、そこで起こっている全ての体細胞突然変異を同定することが可能になってきています。現在こうした技術を用いた大規模ながんゲノム解読プロジェクト(国際がんゲノムコンソーシアム:International Cancer genome Consortium:ICGC(注1)、ならびにがんゲノムアトラス:The Cancer Genome Atlas:TCGA(注4))が開始されています。こうしたプロジェクトの結果から得られる膨大ながんゲノムビッグデータを解析することで、個々のがん種においてどのような体細胞突然変異パターンが起こっており、それぞれがどういった要因によるものか、について解析することが可能になってきました。
国立がん研究センターはこうした国際共同研究ネットワークに参加し、世界の研究者・研究機関とともにがんの解明に取り組んでおり、特に、肝臓がんのゲノムデータの収集と蓄積に主要な役割を担っています。肝臓がんは、日本や中国を含む東アジアとアフリカで発症頻度が高く、世界全体の部位別がん死亡率では第3位に挙げられています。また最近では、アジアだけでなく欧米でも増加しているがんとして世界的に対策が急がれています。
研究手法
今回共同研究グループは、47のがんの専門医療機関および研究機関と共同で、30種類のがんから7,042例のがんゲノムデータを収集し、総計約500万個(4,938,362個)の体細胞突然変異を用いて解析を行いました。12種類の体細胞突然変異を6種類にまとめ(DNAはA:T、C:Gの相補的な2本鎖であるため、例えばC>AとG>Tを区別できない)、さらに体細胞突然変異の周囲の配列情報として直前と直後の塩基(各4種類)を加え、合計で6×4×4=96種類に分類してデータとして用いています。
すでに乳がんの解析で使用しているNon-negative matrix factorization(非負値行列因子分解)(注5)解析手法を用いて、各がん種においてどのような体細胞突然変異パターンの組み合わせがどの程度貢献しているのかについて解析を行いました。さらに突然変異が局所的に増加している領域の抽出を行いました。
主な研究成果
- がんにおける体細胞変異のパターン抽出から、全部で22種類のパターンを発見しました。多くのがん種では2つ以上のパターンの混在が見られ、最大で肝臓がん、胃がん、子宮がんにおいて6種類のパターンの混在が見られました。これらのがんでは多様な発がん要因あるいは分子機構が働いている可能性が考えられます。
- 新たな要因としてDNA変異導入機能を持つAPOBEC遺伝子群の異常によるものが認められました。1.で発見された22種類の中には加齢や喫煙、紫外線といった既知の発がん物質への暴露、既知のDNA修復経路異常(マイクロサテライト不安定性、BRCA1/2異常)、DNA異常を誘導する抗がん剤治療と相関するものが認められましたが、現時点で明らかな要因は同定できないものも多いという結果でした。
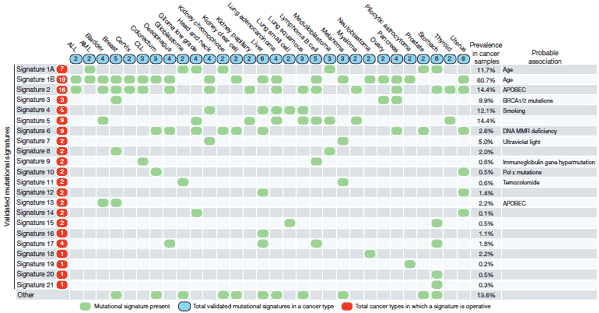
図 さまざまながん種における体細胞変異のパターン
収集した30種類のがん種について、体細胞変異パターンを22種類同定し、それぞれのがんとの関連を分析した。このうち、15のがん種について共通する体細胞変異パターンを有するSignature2ならびにSignature13(上から3行目と14行目)について、APOBEC遺伝子群の異常が新たな発がん要因であることが明らかになった。加齢や喫煙、紫外線といった既知の発がん要因への暴露、DNA修復経路異常(マイクロサテライト不安定性、BRCA1/2異常)、DNA異常を誘導する抗がん剤治療と相関するものもみられた。
今後の期待
今回の研究結果から、発がん要因の異なるさまざまながんにおける突然変異パターンの分類が可能になりました。今後、発がん物質への暴露に関する疫学データや発がんモデル動物を活用することによって、それぞれの変異パターンが誘導される原因を解明し、これまで不明であった多くのがんにおける発がん要因を推定していくことが可能になると考えられます。従って本解析結果は、科学的エビデンスに基づくがん予防のための基礎的な知識基盤となることが期待されます。
また、現在進められている大型がんゲノムプロジェクトから、今後大量のがんゲノムデータが産生されることが考えられます。さらに国立がん研究センターにおいては、既知の治療関連遺伝子変異情報に基づくがん個別化医療の実現に向けた試みとして、東病院において「切除不能・進行・再発固形がんに対するがん関連遺伝子変異のプロファイリングと分子標的薬耐性機構の解明のための網羅的体細胞変異検索(ABC study:Analyses of Biopsy Samples for Cancer Genomics)」、中央病院では「がん患者の臨床検体を用いた、治療効果および毒性に関する遺伝子のプロファイリング研究(TOP-GEAR:Trial of Onco-Panel for Geneprofiling to Estimate both Adverse events and Response by cancer treatment)」が開始されており、こうした研究からは治療反応性等の臨床情報を伴う大量のゲノムデータの収集と集積が進むことが期待されます。
このようながんゲノムビックデータと疫学情報あるいは臨床情報を組み合わせることで、疾患の原因究明に関する未知の発見から発がん経路の解明研究、あるいはがん個別化医療において有用なゲノム異常の同定研究が進展することが期待されます。本研究はそうしたビッグデータ解析の好例であり、今後もこうした解析が当センターを含めた国際的な共同体制のもとで行われることが期待されます。
原論文情報
問い合わせ先・報道担当
問い合わせ先
独立行政法人国立がん研究センター 研究所
がんゲノミクス研究分野
分野長 柴田 龍弘(しばた たつひろ)
Eメール:tashibata●ncc.go.jp(●を@に置き換えください)
電話番号:03-3542-2511(内線3123)
ファクス番号:03-3547-5137
報道担当
独立行政法人国立がん研究センター 広報企画室
電話番号:03-3542-2511(代表)
ファクス番号:03-3542-2545
Eメール:ncc-admin●ncc.go.jp(●を@に置き換えください)
補足説明
注1 国際がんゲノムコンソーシアム(ICGC:International Cancer Genome Consortium)
50種類のがんについて、ゲノム解読データベースを作製し世界の研究者に公開することを目的として、2008年の発足した国際的ながんゲノム研究共同体。現在15の国と地域が参加しており、53のプロジェクトが進行中である。日本からは国立がん研究センターならびに理化学研究所が共同で肝炎関連肝臓がんの解読で参加・貢献している。
注2 高速シークエンサー(次世代型シーケンサー)
2003年のヒトゲノム計画終了後、ヒトゲノムの配列30億塩基対を1,000ドル以下のコストで解読すべく、欧米の政府や企業は技術開発を行ってきた。通常のサンガーシーケンス法と比べて、超大量のDNAシーケンス反応を並列して行う技術であり、現在の第2世代の場合、12日間で約6,000億個の塩基配列を解読することができる(ヒトゲノム6人分をカバー)。さらに、現在開発中の第3世代の場合、高速で一分子シーケンスも可能であり、1日で個人のゲノム解読が可能になりつつある。
注3 APOBEC遺伝子
APOlipoprotein B mRNA Editing enzyme, Catalytic polypeptide-likeの略。DNAの変異導入機能を持つ酵素として機能し、ウイルス感染などに対する宿主防衛機能として発現が誘導されることが知られている。今回の結果からがんゲノムにおける領域的な体細胞変異の集中にも関与している可能性が推測された。
注4 がんゲノムアトラス (The Cancer Genome Atlas)
2005年から国立がん研究所(NCI)ならびに国立ヒトゲノム研究所(NHGRI)が共同で支援している米国のがんゲノム解読プロジェクト。現在20種類以上のがん種について解析を進めている。
注5 Non-negative matrix factorization: 非負値行列因子分解
0か正の値(非負値)のみからなるデータ(行列)を解析する手法。非負値行列を2つの非負値行列に分解するシンプルな解析アルゴリズムであるが、音響信号、画像データ、文書データの解析といったさまざまな分野への応用が可能である。例えば顔画像からの各パーツの特徴抽出の論文(下記1)で使用され、その有効性が注目されている。がんにおける体細胞変異における特徴的なパターン抽出という目的においても有用であることが今回の検討で示された。
- D. D. Lee and H. S. Seung, Learning the parts of objects by non-negative matrix factorization, Nature, Vol. 401, No. 6755, pp. 788-791, 1999.