世界規模の国際ネットワークによる最大のがん種横断的全ゲノム解読日本人症例での解析を進めることで日本人に最適な臨床開発への発展を期待
2020年2月6日
国立研究開発法人国立がん研究センター
in English
発表のポイント
-
37カ国1,300名を超える世界中のがん研究者が参加した国際共同研究によって、過去最大の38種類のがん、2,658症例のがん全ゲノム解読データが統合解析されました。
-
これまで明らかではなかったヒトゲノムの約99%を占める非コード領域 注1における新たなドライバー異常、突然変異や染色体構造異常に見られる特徴的なパターンの解明など、ヒトがんゲノムの多様な全体像が明らかになりました。
-
希少がんなど約5%の症例ではドライバー変異が特定されなかったため、がんドライバーの発見はまだ完了していないことが示唆されました。
-
研究成果はデータポータルで公開され、世界中のがん研究者が活用できます。
-
今後、日本人症例での全ゲノム解析なども行うことで、日本人に最適な臨床開発へ発展することが期待できます。
概要
国立研究開発法人国立がん研究センター(理事長:中釜 斉、東京都中央区)は、国際がんゲノムコンソーシアム(International Cancer Genome Consortium: ICGC) 注2が主導するがん種横断的な全ゲノム解析プロジェクト(Pan-Cancer Analysis of Whole Genome: PCAWG)に参加し、これまでで最大の38種類のがん2,658症例のがん全ゲノム解読データを統合解析した研究成果を科学誌「Nature」で6本の論文として英国時間2月5日付け(日本時間2月6日)に発表しました。過去に例のない巨大ながん全ゲノム解読データによって、これまで明らかではなかったヒトゲノムの約99%を占める非遺伝子領域における新たな異常や、突然変異や染色体構造異常に見られる特徴的なパターンの解明など、ヒトがんゲノムの多様な全体像が詳細に解明されました。
一方で、がんにおけるドライバー遺伝子や変異パターンには人種による違いがあることがこれまでの研究から知られていますが、今回の解析では日本人症例のサンプル数が286例と十分でなく、また胃がん・食道扁平上皮がん・胆道がんなどアジア・日本に多い難治がんについては症例数が少ないため特徴を見出すには至りませんでした。今後、日本人のがん症例について大規模な全ゲノム解析を行い、そのデータと精度の高い臨床情報とを合わせたビックデータが構築されることで、全ゲノム情報の活用による新たな臨床開発が進むことも期待されます。
背景
がんはゲノム(遺伝子全体)の異常によって引き起こされる疾患であり、その異常を知ることはがんの診断・治療・予防にとって重要な情報となるだけでなく、新たな治療薬の開発によっても不可欠です。近年いわゆる次世代シークエンサーの出現により、がん細胞におけるゲノム異常を全ゲノム規模で解析することが可能になってきました。
これまでのがんゲノム研究の多くは、タンパク質をコードしている領域(ゲノム全体の約1%を占める)における異常に注目して解析を行っています。その成果は、分子標的治療開発や今まさに日本でも開始されているがんゲノム医療という形で実用化され、がん患者さんの治療・診断に活用されています。一方でゲノムの残り約99%の非コード領域における異常については、その意味が十分に理解されておらず、診断や治療への活用もほとんど進められていません。とりわけ非コード領域における異常の意義を理解するためには、多数のサンプルについて全ゲノム解読を行い、また遺伝子発現など他の分子情報と統合して解析する必要があるため、その全貌を明らかにすることは困難でした。
そこで、2008年に発足したがんゲノム研究に関する世界最大の国際共同研究(国際がんゲノムコンソーシアム)では、コンソーシアム内で集積した38種類のがん、2,658症例のがん全ゲノム解読データについて、1,300名を超える世界中のがん研究者(日本からは60名以上)が参加のネットワークによって、様々な視点から解析を行い、がんゲノムの理解を進めるプロジェクト(Pan-Cancer Analysis of Whole Genome: PCAWG: がん種横断的な全ゲノム解析プロジェクト)を2015年から開始しました。これは現時点で世界最大のがん全ゲノム解読の研究です。
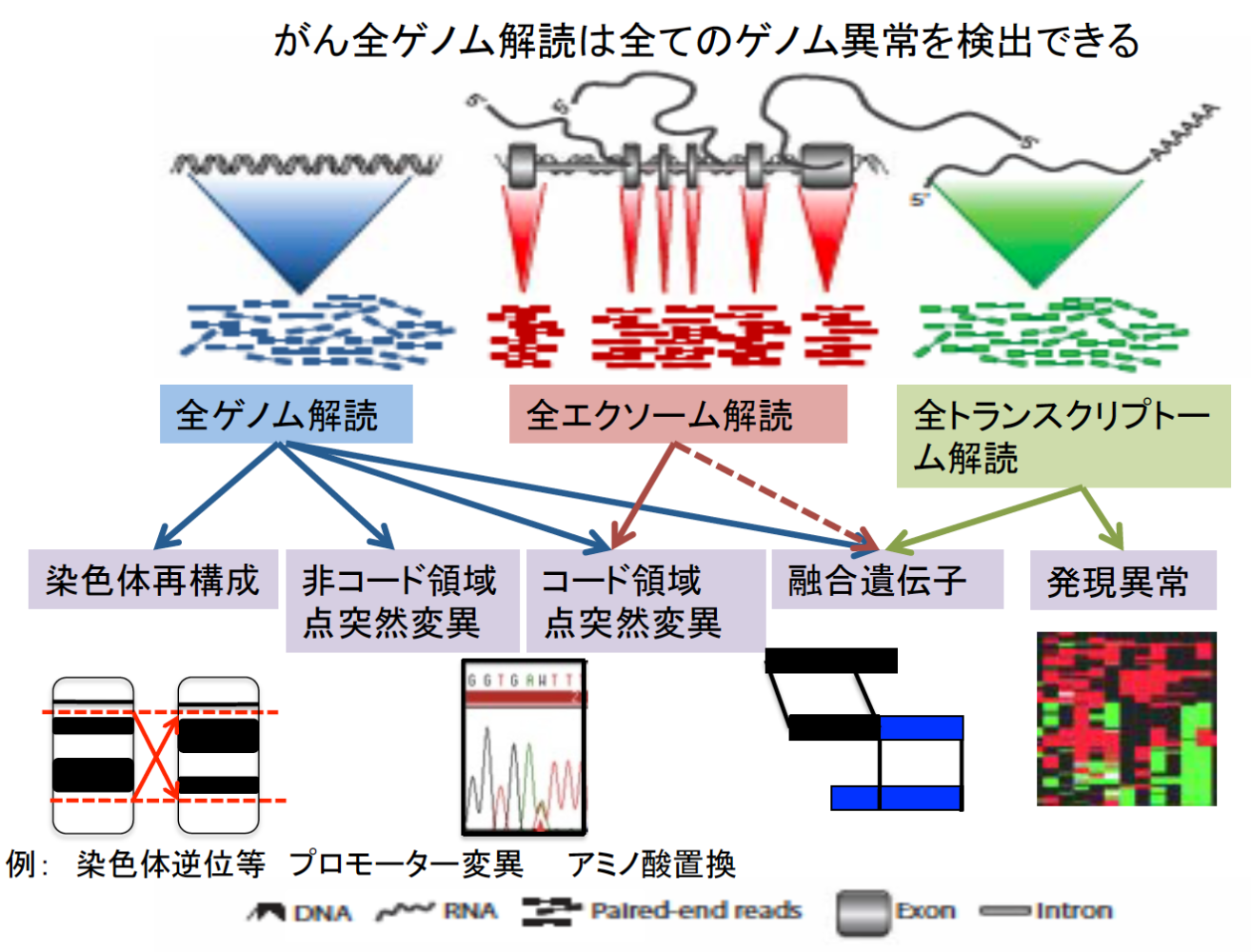
研究方法
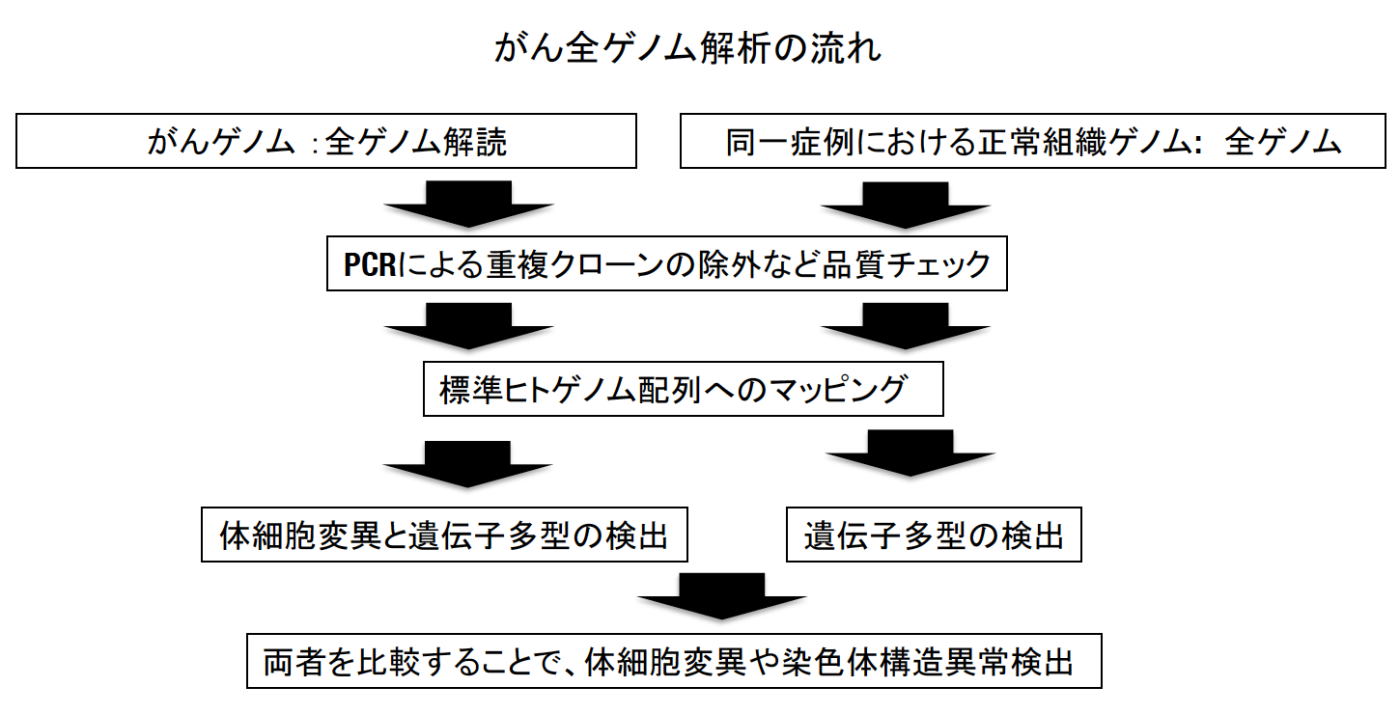
得られたシークエンスデータは、品質検定後、全て統一された解析パイプライン(今回は3つの解析パイプライン注3の結果を参照し、より精度の高いデータを産出した)によって、体細胞変異・染色体構造異常・コピー数異常を検出しました(下図)。さらに、その結果について、それぞれテーマの異なる16の解析グループ(下記参照)が様々な視点から解析を行いました。
解析テーマの一部
-
非コード領域を含めたがんにおけるドライバー遺伝子の同定とその機能的解釈
-
ゲノム異常が遺伝子発現に与える影響について
-
がんゲノムにおける染色体構造異常の同定とその特徴の解明
-
変異シグネチャーの同定と発がん要因暴露との関連
-
生殖細胞系列多型ががんゲノムに与える影響について
-
がんゲノムの臨床病理学的意義の理解
-
がんゲノム進化の研究
-
免疫ゲノム並びにミトコンドリアゲノムにおける異常
-
病原体ゲノムの同定とその意義の理解
主な研究成果
全てのデータはデータポータル(https://dcc.icgc.org/pcawg 外部サイトにリンクします)上で公開され、世界のがん研究者が活用できます。
-
がんゲノムには、タンパク質コード領域および非コード領域を合わせて平均して4~5個のドライバー変異が含まれていました。しかし、希少がんなど一部の症例の約5%ではドライバー変異が特定されなかったため、がんドライバーの発見はまだ完了していないことが示唆されました。
-
単一の破壊的事象によって多くの染色体構造異常が局所的に集中して発生するクロモスリプシス(染色体粉砕)は、しばしば腫瘍進化の初期事象として認められます。例えば、末端性黒色腫では、これらの事象はほとんどの体細胞点変異より先に起こり、いくつかのがん関連遺伝子に同時に影響を及ぼします。
-
テロメア注4の維持異常を伴うがんは、複製活性が低い組織から生じることが多く、テロメアの臨界レベルまでの短縮を防ぐいくつかの分子機構を呈しています。生殖系列遺伝多型は、高頻度あるいはまれな多型のいずれも、点変異、染色体構造異常、レトロトランスポゾン転位注5など、体細胞変異のパターンに影響を及ぼします。
-
TERTプロモーター注6における非コード領域変異以外にも、少数ではあるががんを引き起こす非コード領域変異があることが明らかになりました。
-
塩基置換、小さな挿入や欠失、染色体構造異常を引き起こす変異誘発過程における新しいシグネチャーを同定しました。
-
スプライシング・発現レベル・融合遺伝子・プロモーター活性といった点において体細胞変異が転写に様々な影響を及ぼすことを複数のがん種において解明しました。
展望
本研究プロジェクトは、がんという人類が克服すべき疾患に対して、広く世界中のがん研究者が国際協力し、2,600症例を超える世界最大のがんゲノムデータの解析を行った地球規模での金字塔的な研究成果です。この研究によって、これまで未解明であったゲノム暗黒領域(ダークマター)と呼ばれる非コード領域におけるがんゲノム異常の意味が、全てではありませんが、明らかにされました。今後本研究成果から、がんという病態の理解と同時に、新たな診断法や治療法の開発が進むことが期待されます。また将来的には、現在ゲノム医療で用いられている遺伝子パネルの更新にもその成果が活用されることが期待されます。
一方で約5%の症例では明らかなドライバー遺伝子を同定できませんでした。この理由の一つは、がん種によっては症例数が少ないため、稀なドライバー遺伝子を検出する統計的パワーが十分ではなかった可能性が考えられ、更に各がん種について多数症例のデータを集積する必要があると考えられます。またこれまでの研究から、がんにおけるドライバー遺伝子や変異パターンには人種による違いがあることが知られていますが、今回の解析では日本人症例における特徴を見出すにはサンプル数が十分ではありませんでした。今後、わが国におけるゲノム医療を最適化していくためには、日本人がん症例について更に大規模な全ゲノム解析を行うことが必要であると考えられます。またこうした全ゲノムデータと精度の高い臨床情報と合わせたビックデータを構築することで、全ゲノム情報の活用による新たな臨床開発が進むことも期待されます。
また本プロジェクトでは、がん全ゲノム解析手法の開発や標準化、クラウドによるがんゲノム解析、更に発現情報やエピゲノムデータとの統合解析など、次世代のがんゲノム解析フレームワークを提供したという点でも大きな意義があります。
現在国際がんゲノムコンソーシアムでは、新たなプロジェクトとして、より大規模で豊富な臨床データを付加した200,000症例のがん全ゲノム解読データを集積する後継プロジェクトであるICGC-ARGO注7を開始し、当センターも日本の代表機関として引き続き参加を表明しています。今回の研究成果は、大規模ながん全ゲノム解析研究を推進する上で必要な情報解析基盤を構築し、その実現可能性を示すと同時に、今後のがんゲノム研究の趨勢・方向性に大きな影響を与えるものと考えます。
発表論文
雑誌名: Nature
タイトル: 合計6本の論文がNature誌に同時掲載。(下記1~3について国立がん研究センターの研究者の貢献が大きいものです。)
-
Pan-cancer analysis of whole genomes.(外部サイトにリンクします)
-
The Repertoire of Mutational Signatures in Human Cancer.(外部サイトにリンクします)
-
Genomic basis of RNA alterations in cancer.(外部サイトにリンクします)
-
Patterns of somatic structural variation in human cancer genomes.(外部サイトにリンクします)
-
Analyses of non-coding somatic drivers in 2,693 cancer whole genomes.(外部サイトにリンクします)
-
The evolutionary history of 2,658 cancers.(外部サイトにリンクします)
著者: The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium
研究費
国立研究開発法人日本医療研究開発機構
革新的がん医療実用化研究事業
「国際共同研究に資する日本人難治性がん・生活習慣病関連がん大規模統合ゲノミクス解析と国際コンソーシアムでのデータ共有による国際貢献」
国立がん研究センター研究開発費29-A6
「がん情報生物学・生物統計学研究基盤の構築」
用語解説
注1 非コード領域
酵素など細胞の様々な機能に携わるタンパク質の配列(遺伝子)がDNAに書き込まれた領域をコード領域 (coding region)と呼び、ヒトゲノムの約1%を占める。一方で残りの約99%の領域は、非コード領域(non-coding region)と呼ばれ、遺伝子発現の制御領域(プロモーターやエンハンサー)、制御RNA (transfer RNA, ribosomal RNA, microRNA、lincRNAなど)、あるいはテロメアやセントロメアなど染色体構造維持に必要な領域や多くの反復配列が含まれる。がん細胞ではこうした領域にも様々な異常が起こっているが、これまでその意義は十分に解明されていなかった。
注2 国際がんゲノムコンソーシアム
(https://icgc.org 外部サイトにリンクします)
国際共同がんゲノムプロジェクト「国際がんゲノムコンソーシアム」(International Cancer Genome Consortium:ICGC)は、世界各国を通じて臨床的に重要ながんを選定し、国際協力で包括的かつ高解像度のゲノム解析を行い、がんのゲノム異常の包括的カタログを作成し、網羅的がんゲノム情報を研究者間で共有および無償で公開することでがんの研究および治療を推進することを目的に2008年に発足しました。現在、17カ国が参画し、 総計90に及ぶ様々ながん種についての大規模ゲノム研究プロジェクトが精力的に遂行されています。国立がん研究センターは日本の代表研究機関として肝臓がん・胆道がん・胃がんの解析を担当している。
注3 解析パイプライン
次世代シークエンサーによって得られた大量の配列情報から、大型計算機やクラウドを使ってがん全ゲノムデータを解析するためのワークフロー。がんゲノムにおける異常を検出するためには、まずがん組織並びに同じ患者さんの正常組織の全ゲノム配列を標準ヒトゲノム配列(ヒトゲノム計画によって作成された配列。レファレンスゲノムとの呼ばれる)と比較し、その違いを検出し、更にがん組織と正常組織の違いを見つけることで、がん組織にのみ起こっている異常(体細胞ゲノム異常)を同定する。今回のプロジェクトではパイプラインによる影響をなるべく除外するために、3つの異なるパイプラインで同じデータを解析し、全てのパイプラインで完全一致したものを主に用いた。
注4 テロメア
染色体の末端にある構造で、染色体末端を保護する機能を持つ。テロメアが失われると、染色体融合といった構造異常を誘発する。正常細胞は分裂するたびにテロメアがわずかに短縮し、ある一定長以上より短くなると増殖を停止し、細胞老化を引き起こす。しかしがん細胞のように細胞分裂が盛んな細胞では、テロメアを伸長させる酵素であるテロメラーゼの発現が上昇するなど様々な分子機構によってテロメアの短縮を防いでいる。
注5 レトロトランスポゾン転位
レトロトランスポゾン (Retrotransposon) は、可動遺伝因子(トランスポゾン)の一種であり、レトロトランスポゾンは、自分自身を RNA に複写した後、逆転写酵素によって DNA コピーを作成し、それをゲノムの他の部分に挿入することが知られており、これを「転位(transposition)」と呼ぶ。LINE(長い散在反復配列)やSINE(短い散在反復配列)を合わせて、ヒトゲノムの30%以上がレトロトランスポゾンで構成されている。がん細胞では、一部のレトロトランスポゾンが特異的に転位することが知られており、ゲノム不安定性やがん抑制遺伝子の不活性化に関与していると報告されている。
注6 TERTプロモーター
テロメアを伸長させる酵素であるテロメラーゼ(TERT)遺伝子の発現を制御領域する領域。正常組織ではTERT遺伝子の発現は、幹細胞などごく限られた細胞に限定するように制御されている。脳腫瘍、メラノーマ、甲状腺がん、肝臓がん、膀胱がんなどがんにおいて、高頻度にこの制御領域の点変異が起こることが知られている。変異の結果、本来は結合できないはずのETSという転写因子が結合できる配列が作られ、その結果がん細胞でTERT遺伝子の発現量が増加する。
注7 ICGC-ARGO (International Cancer Genome Consortium- Accelerating Research in Genomic Oncology)
(https://www.icgc-argo.org 外部サイトにリンクします)
ICGCにおける次期プロジェクト。ICGCやPCAWGプロジェクトの後継として、200,000症例のがんについて全ゲノムデータを集積し、同時に豊富な臨床病理情報を付加することによって、革新的な治療・予防法の開発や、治療抵抗性の分子機構の解明などを目指すプロジェクト。現在日本を始め、米国・カナダ・英国・ドイツ・フランス・イタリア・スイス・韓国・中国・香港・サウジアラビアの12カ国が参加を表明している。
お問い合わせ先
国立研究開発法人国立がん研究センター
企画戦略局 広報企画室
〒104-0045 東京都中央区築地5-1-1
電話番号:03-3542-2511(代表) FAX:03-3542-2545 Eメール: ncc-admin●ncc.go.jp(●を@に置き換えてください)
関連ファイル
PDFファイルをご覧いただくには、Adobe Readerが必要です。Adobe Readerをお持ちでない方は、バナーのリンク先から無料ダウンロードしてください。