トップページ > 研究組織一覧 > 分野・独立ユニットグループ > がん治療学研究分野 > 研究成果の概要 > グルタチオン代謝脆弱性
グルタチオン代謝脆弱性
メインテーマ概要
グルタチオン代謝脆弱性とは
本ページでは、SWI/SNF クロマチンリモデリング複合体の構成因子(ARID1A・SMARCB1 など)が欠損したがんに共通して生じる「グルタチオン代謝の綻び」を治療標的化する 研究成果を紹介します。
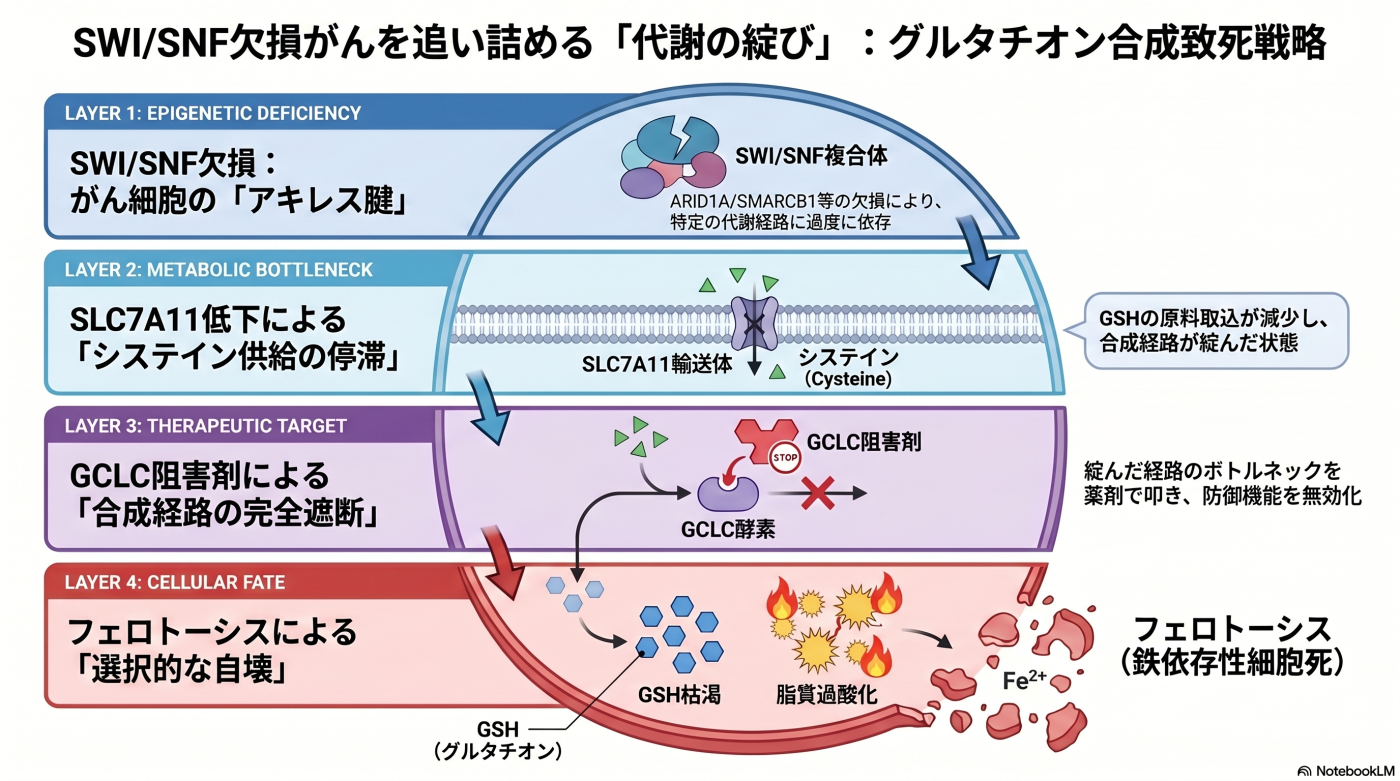
グルタチオン代謝脆弱性 は、「エピゲノム異常がもたらす代謝の綻び」を標的とする合成致死戦略です。SWI/SNF 複合体の構成因子が欠損したがん細胞では、シスチントランスポーター SLC7A11 の転写が低下し、抗酸化システムの中心を担う グルタチオン(GSH) の合成原料(システイン)の供給が慢性的に不足する状態となります。この 既に綻んだ代謝経路 をさらに追い込むことで、がん細胞選択的な治療効果を得ることを目指します。
私たちは、この概念を 2019 年から2020年に ARID1A 欠損がんで報告(Ogiwara et al., Cancer Cell, 2019)(Sasaki et al., Biochem Biophys Res Commun. 2020)し、その後、SWI/SNF 複合体欠損がん全般への普遍化(Sasaki et al., Sci Rep, 2024)、SMARCB1 欠損希少がんへの深化と新規 GCLC 阻害剤の創製(Takeuchi et al., Cancer Res, 2026)と、段階的に研究を展開してきました。
研究室の研究系譜での位置づけ
本テーマは、研究室の合成致死戦略における 代謝脆弱性アプローチを代表する研究系譜 です。2019 年の Cancer Cell 論文で ARID1A 欠損がんにおける GSH 代謝依存性を報告し、その後、SWI/SNF 欠損がん全般への普遍化、SMARCB1 欠損希少がんに対する新規 GCLC 阻害剤の創製(国立がん研究センターと小野薬品工業株式会社との共同研究 によりフェロトーシスを介した作用機序の解析)へと展開してきました。
本テーマは、研究室の他の主軸 ── 従来型合成致死(1 対 1 対応型)、パラログ同時阻害法(1 対 2 対応型)、次世代型合成致死(2 対 n 対応型)、データ駆動型探索 ── と並行する 代謝脆弱性軸の研究 として位置づけられます。
このページに掲載する研究
- サブテーマ 1: ARID1A 欠損がん × GCLC/GSH 代謝 ── Ogiwara et al., Cancer Cell, 2019、Sasaki et al., Biochem Biophys Res Commun. 2020。本テーマの 起点となる研究。「エピゲノム × 代謝」の交差点を治療標的化する設計思想を確立
- サブテーマ 2: SMARCB1 欠損希少がん × 新規 GCLC 阻害剤 GCLCi1/GCLCi0 ── Takeuchi et al., Cancer Res, 2026。小野薬品工業株式会社との共同研究。フェロトーシス機構の解明と GLS 阻害剤との合理的併用療法
- サブテーマ 3: SWI/SNF 欠損がん全般への普遍化 ── Sasaki et al., Sci Rep, 2024。ARID1A、SMARCA4、SMARCB1、PBRM1 等の SWI/SNF 構成因子欠損がん全般に共通する代謝脆弱性の証明
サブテーマ 1: ARID1A 欠損がん × GCLC/GSH 代謝 ── 代謝脆弱性研究の起点
研究要約
|
項目 |
内容 |
|---|---|
|
対象がん |
ARID1A 欠損がん: 卵巣明細胞がん(OCCC、約 50%)、びまん性胃がん(約 25%)、子宮体がん(約 20%)等 |
|
遺伝子背景 |
ARID1A 欠損 ── SWI/SNF クロマチンリモデリング複合体(cBAF サブタイプ)の構成因子の機能喪失 |
|
標的・経路 |
グルタチオン(GSH)代謝経路/SLC7A11(シスチントランスポーター)/GCLC(GSH 合成律速酵素)/GPX4(GSH 依存性脂質過酸化抑制酵素) |
|
作用機序 |
ARID1A 欠損 → SLC7A11 転写低下 → シスチン取り込み減少 → 細胞内システイン供給不足 → GSH 合成が綻んだ状態 → GSH 阻害剤(Eprenetapopt、BSO 等)への高感受性 → 酸化ストレス過剰 → 細胞死誘導 |
|
創薬上の意義 |
ARID1A 欠損がんが GSH 代謝に依存することを 世界に先駆けて報告。「エピゲノム × 代謝」の交差点を治療標的化する独自の設計思想を確立。研究室の代謝脆弱性研究の起点となる成果 |
|
検証段階 |
網羅的代謝経路スクリーニング/in vitro(ARID1A 欠損 vs 保持での選択的応答)/in vivo(マウス xenograft モデルでの腫瘍縮小)/SLC7A11 の強制発現実験による機構検証 |
|
創薬段階 |
研究ツール化合物 Eprenetapopt、l-buthionine sulfoximine(BSO) を用いた概念実証段階/後のサブテーマ 2 で新規 GCLC 阻害剤の創製につながった |
|
代表論文 |
Ogiwara et al., Cancer Cell, 2019、 Sasaki et al., Biochem Biophys Res Commun. 2020 |
このサブテーマの要点
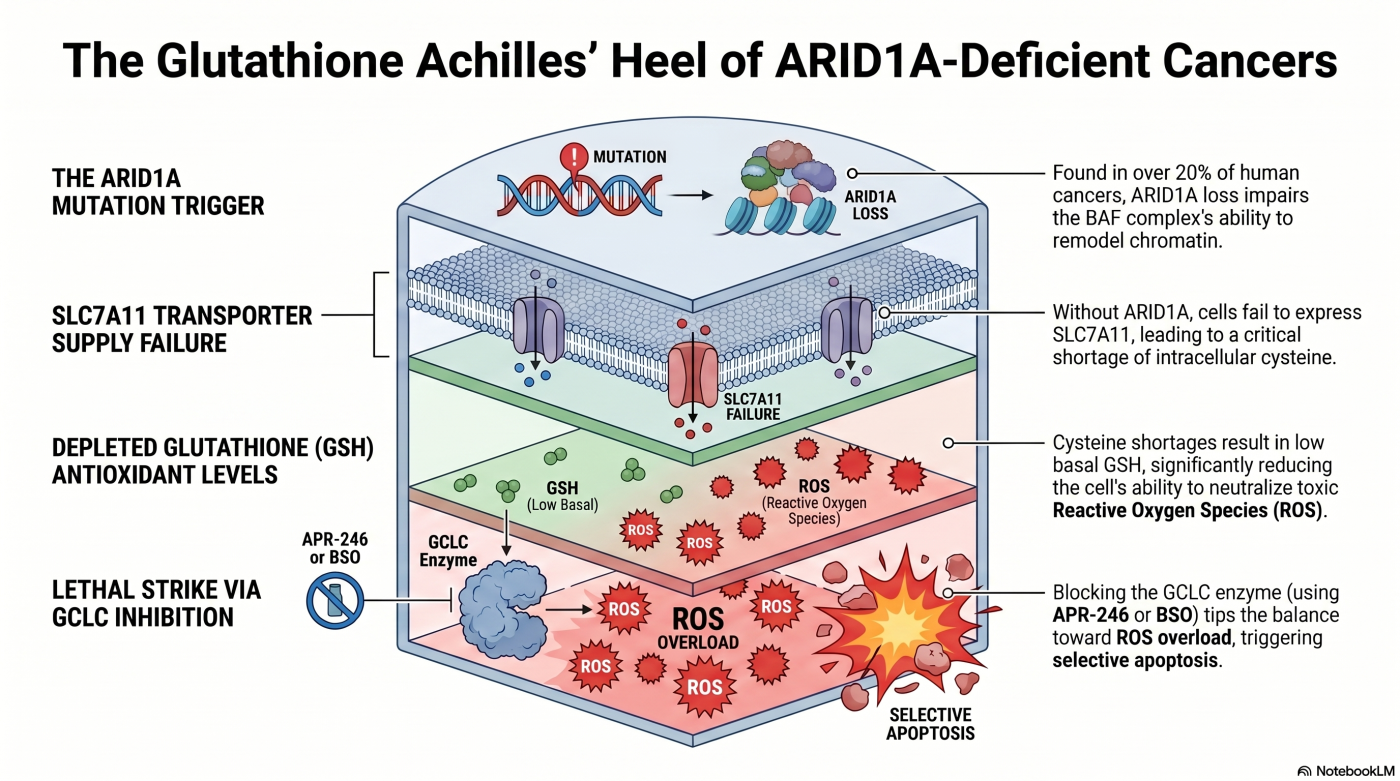
私たちは、独自の細胞株パネルを用いた網羅的な代謝経路スクリーニングにより、ARID1A 欠損がん細胞がグルタチオン(GSH)代謝に強く依存している ことを示しました(Ogiwara et al., Cancer Cell, 2019)。
中核となる機序は、ARID1A が シスチントランスポーター SLC7A11 のプロモーター領域に結合してその転写を維持する役割を担う ことを発見した点です。ARID1A が欠損すると、SLC7A11 の発現が低下し、細胞外からのシスチン取り込みが減少します。シスチンは GSH 合成の必須基質である システイン に還元される前駆体であるため、その供給不足は GSH 合成が既に綻んだ状態 を生み出します。
このため、ARID1A 欠損がん細胞は、GSH 枯渇を誘導する薬剤(Eprenetapopt、BSO 等)に対して正常細胞よりはるかに高い感受性を示します。本研究は、エピゲノム制御因子の欠損が 代謝経路の特定のノードを脆弱化する という、研究室の代謝脆弱性アプローチの 起点となった成果 です。
背景
卵巣明細胞がん(OCCC)は、卵巣がんの組織型のうち日本人女性に比較的多いサブタイプであり、プラチナ製剤を中心とする標準化学療法が効きにくい ことが長年の課題でした。再発例では治療選択肢が限られ、新しい治療標的の発見が強く求められてきました。ARID1A は、卵巣明細胞がんの約半数で機能喪失型変異が認められる、最も高頻度のドライバー遺伝子の一つです。
ARID1A 欠損は、OCCC のほか、びまん性胃がん(約 25%)、子宮体がん(約 20%)等、複数のがん種にまたがって認められます。これら ARID1A 欠損がんに共通する難しさは、ARID1A が「がん抑制遺伝子」(機能喪失型変異)であり、そのもの自体を直接標的にすることが原理的にできない という点にあります。
私たちは、この困難を乗り越えるアプローチとして、「失われた遺伝子の代わりに、失われたことで生まれた『代謝の綻び』を突く」 という戦略を選びました。これが、本研究系譜の出発点です。
発見
網羅的代謝経路スクリーニング
私たちは、独自の細胞株パネル(ARID1A 欠損型 と ARID1A 保持型)に対して、網羅的な代謝経路スクリーニング を実施しました。その結果、ARID1A 欠損細胞株が グルタチオン(GSH)関連経路に共通して脆弱性を示す ことを同定しました。
ARID1A による SLC7A11 転写制御の発見
中核となる発見は、ARID1A が シスチントランスポーター SLC7A11 のプロモーター領域に結合し、その転写を維持する役割を担っている という機序の解明です。ARID1A 欠損下では、SLC7A11 の転写が低下し、細胞外からのシスチン取り込みが減少します。
取り込まれたシスチンは、細胞内で還元されて システイン となり、GSH 合成の必須基質 として消費されます。SLC7A11 の発現低下は、システイン供給の慢性的な不足を意味し、その結果、ARID1A 欠損がん細胞では GSH 合成が既に綻んだ状態 にあります。
作用機序
ARID1A 欠損がん細胞における GSH 合成の綻びは、以下の連続的な現象として機能します。
- ARID1A 欠損: SLC7A11 プロモーター領域への結合喪失
- SLC7A11 転写低下: シスチン取り込み能力の減少
- システイン供給不足: GSH 合成の必須基質の慢性的不足
- GSH 合成のボトルネック: 細胞内 GSH 量が低めに維持される
この状態のがん細胞では、GSH を 直接枯渇させる薬剤(Eprenetapopt)や、GSH 合成律速酵素 GCLC を阻害する薬剤(BSO)に対して、正常細胞よりはるかに高い感受性を示します。一方、ARID1A が保たれている細胞では SLC7A11 発現が維持され、GSH 合成能力にも余裕があるため、同等の薬剤投与でも顕著な細胞死は誘導されません。これが、がん細胞選択性が得られる可能性 の理論的根拠となります。
ARID1A 欠損そのものを薬理学的に回復させることは困難ですが、その代償として生じる「代謝の綻び」は治療標的になり得ます ── これが、本研究で明らかになった合成致死性の生物学的本質です。
検証
細胞株モデル(in vitro)
ARID1A 欠損 vs 保持の細胞株パネルに対する GSH 阻害剤 Eprenetapopt や GCLC 阻害剤 BSO の投与により、ARID1A 欠損細胞株は ARID1A 保持細胞株に比べて 顕著に高い感受性 を示しました。
マウス xenograft モデル(in vivo)
ARID1A 欠損 OCCC 細胞株を用いたマウス異種移植モデルにおいて、Eprenetapopt 単剤投与で腫瘍縮小効果 が確認されました。
機構の因果検証
SLC7A11 を ARID1A 欠損細胞に 強制発現 させると、GSH 標的薬剤に対する感受性が解除されました。これは、SLC7A11 が ARID1A 欠損 → GSH 依存性の中核メディエーター であることを支持する重要な所見です。また、ARID1A 欠損がん細胞での GSH レベルの実測、SLC7A11 mRNA/タンパク質量の定量、酸化ストレス指標の評価など、複数の独立した実験系で機構を検証しています。
創薬・臨床への展望
本研究は、ARID1A 欠損がんに対する代謝脆弱性アプローチ の科学的基盤を提供するものです。想定される対象患者層は、ARID1A 欠損 OCCC(卵巣がんに占める割合が日本人女性で比較的高い)、ARID1A 欠損びまん性胃がん、ARID1A 欠損子宮体がん 等の患者さん となります。
ARID1A 欠損 という明確な遺伝子バイオマーカーに基づき、「効く可能性が高い患者さんをあらかじめ選別し、GSH 代謝阻害剤を含む代謝標的治療法の開発につなげる」 ── これが、本研究が目指す個別化医療(プレシジョン・メディシン)の枠組みです。
本研究で用いた Eprenetapopt・BSO は研究ツール化合物としての位置づけであり、本研究の意義は、ARID1A 欠損がんに対する GSH 代謝経路の脆弱性を概念実証 した点にあります。この概念実証を基盤として、研究室では本テーマを SWI/SNF 欠損がん全般への普遍化(サブテーマ 3)、SMARCB1 欠損希少がんへの深化と新規 GCLC 阻害剤の創製(サブテーマ 2)へと展開してきました。
関連論文
- Ogiwara H*, Takahashi K, Sasaki M, Kuroda T, Yoshida H, Watanabe R, Maruyama A, Makinoshima H, Chiwaki F, Sasaki H, Kato T, Okamoto A, Kohno T*. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell. 2019;35(2):177–190.e8. PubMed → ARID1A 欠損がんがグルタチオン代謝に依存することを示し、SLC7A11 を介した「エピゲノム × 代謝」の交差点を治療標的化する設計思想を確立。研究室の代謝脆弱性研究の起点。PI(荻原)責任著者論文。
- Sasaki M, Chiwaki F, Kuroda T, Komatsu M, Matsusaki K, Kohno T, Sasaki H, Ogiwara H*. Glutathione Synthetic Inhibition Restores the Sensitivity of Cancer Cells with Epigenetic Repressor Mutations to Cisplatin and Anti-PD-1 Therapy. Biochem Biophys Res Commun. 2020;522(2):342–347. PubMed → びまん性胃がんにおけるグルタチオン合成阻害の感受性を検証した成果。Cancer Cell 2019 の知見を胃がんモデルに展開。
関連プレスリリース
- 2019 年 1 月 25 日: 日本人に多い卵巣明細胞がんなどでみられる ARID1A 遺伝子変異がんを対象に、メタボロームを標的とした新たながん治療法を発見 → プレスリリース
サブテーマ 2: SMARCB1 欠損希少がん × 新規 GCLC 阻害剤 GCLCi1/GCLCi0 ── フェロトーシス機構の解明と共同創薬
研究要約
|
項目 |
内容 |
|---|---|
|
対象がん |
SMARCB1 欠損 希少がん: 悪性ラブドイド腫瘍(MRT、ほぼ全例で SMARCB1 欠損、小児がん)、類上皮肉腫(ES、約 90% で SMARCB1 欠損、若年成人がん) |
|
遺伝子背景 |
SMARCB1 欠損 ── SWI/SNF クロマチンリモデリング複合体(cBAF サブタイプ)の必須サブユニットの機能喪失 |
|
標的・経路 |
GCLC(GSH 合成律速酵素)/GPX4/フェロトーシス(鉄依存性細胞死)経路。サブテーマ 1 と共通の SLC7A11 低下 → システイン供給不足 → GSH 合成の綻び を起点 |
|
作用機序 |
サブテーマ 1 と共通の SLC7A11 低下による GSH 合成の綻びに加え、新規 GCLC 阻害剤による GSH 合成の追加遮断 → GPX4 活性低下 → 脂質過酸化 → フェロトーシス(鉄依存性細胞死)誘導 |
|
創薬上の意義 |
SMARCB1 欠損希少がんに対するフェロトーシスを介した新規治療標的化。GLS 阻害剤との合理的併用療法による相乗効果の実証。国立がん研究センターと小野薬品工業株式会社との共同研究成果 |
|
検証段階 |
in vitro(SMARCB1 欠損 vs 保持での選択的細胞死、Ferrostatin-1 によるレスキュー、脂質過酸化マーカー上昇、GSH 実測)/in vivo(MRT 異種移植モデルでの腫瘍増殖抑制、GCLC + GLS 併用での相乗効果) |
|
創薬段階 |
新規 GCLC 阻害剤 GCLCi1/GCLCi0(小野薬品工業との共同研究で創製、論文公開済み)/GLS 阻害剤 Telaglenastat(CB-839)(複数のがん種で臨床試験が行われてきた薬剤)との併用療法 |
|
代表論文 |
このサブテーマの要点
私たちは、SMARCB1 欠損希少がん(悪性ラブドイド腫瘍 および 類上皮肉腫)に焦点を絞り込み、グルタチオン代謝脆弱性のメカニズムを 作用機序のレベルで深く掘り下げる段階 に入りました(Takeuchi et al., Cancer Res, 2026)。
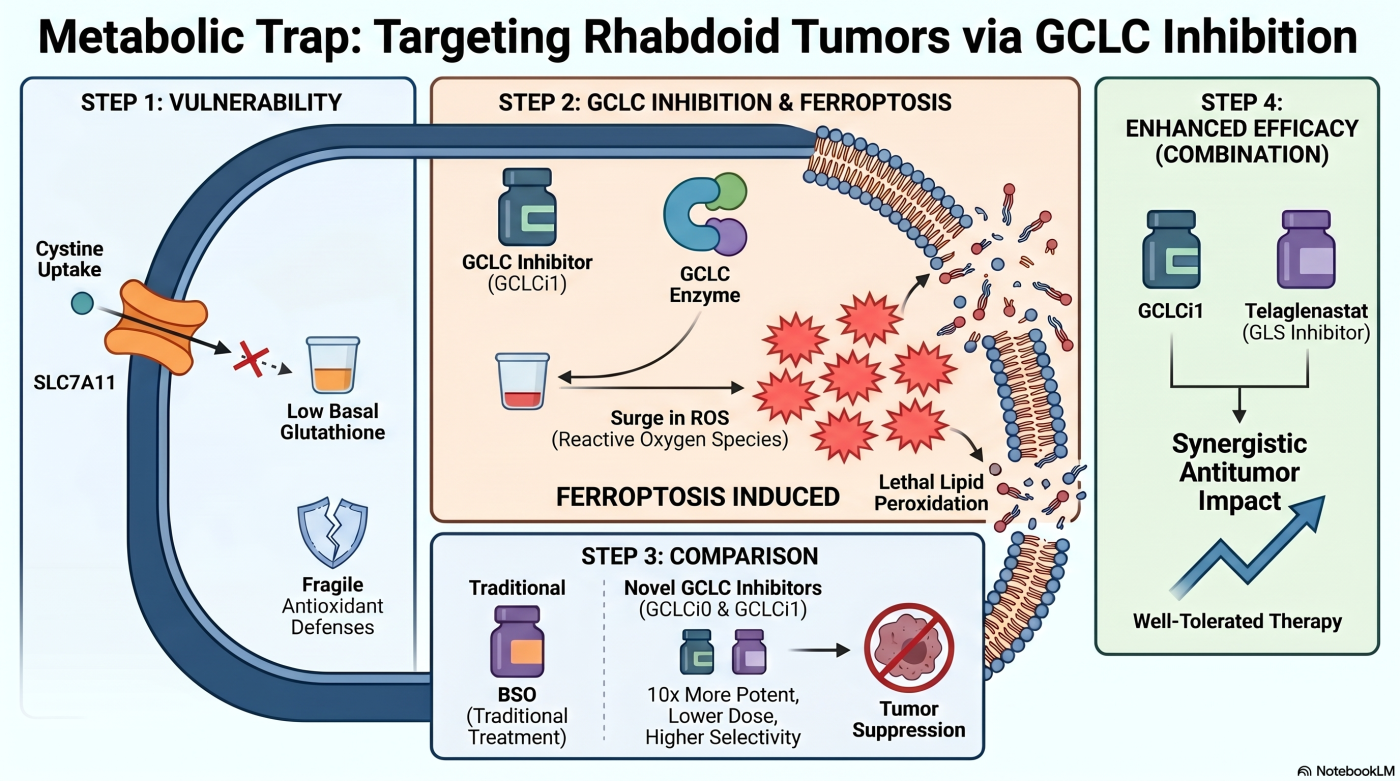
SMARCB1 欠損がんでは、サブテーマ 1 で示した ARID1A 欠損と同様に SLC7A11 の転写が減弱 しており、GSH 合成の必須基質であるシステインの供給が慢性的に不足しています。私たちは、この状態に対して 「GSH 合成の律速酵素である GCLC を直接阻害すれば、GSH 合成の律速段階を直接遮断できる」 と仮説を立て、国立がん研究センターと小野薬品工業株式会社との共同研究 により 新規 GCLC 阻害剤 GCLCi1/GCLCi0 を創製しました。
これらの化合物は SMARCB1 欠損がん細胞において フェロトーシス(鉄依存性細胞死) を介した選択的な細胞死を誘導します。さらに、GLS(グルタミナーゼ)阻害剤 Telaglenastat(CB-839)との合理的併用療法 により、in vivo マウス異種移植モデルで単剤を上回る相乗的な抗腫瘍効果が確認されました。
背景
悪性ラブドイド腫瘍(MRT)は、生後 1 年未満から 3 歳以下の乳幼児に発症する、極めて予後不良な小児固形がん です。脳・腎臓・軟部組織などに発生し、現在の標準治療では長期生存例が限られています。SMARCB1 遺伝子の双アレル性欠損 が、ほぼ全症例で原因として確認されています。
類上皮肉腫(ES)は、若年成人の四肢に好発する、極めて稀な軟部肉腫 です。診断時にはすでに進行例であることが多く、化学療法・放射線療法・外科切除に抵抗性を示すことが知られています。SMARCB1 欠損をターゲットとする EZH2 阻害剤 Tazemetostat が承認されていますが、奏効率や奏効期間に課題があり、新たな治療選択肢が待たれていました。
これらの SMARCB1 欠損がんに共通する難しさは、ARID1A 欠損がんと同様、SMARCB1 が「がん抑制遺伝子」(機能喪失型変異)であり、そのもの自体を直接標的にすることが原理的にできない という点にあります。私たちは、サブテーマ 1 で確立した「代謝の綻びを突く」戦略を、これらの SMARCB1 欠損希少がんに適応拡大しました。
発見
SLC7A11 転写減弱とシステイン供給不足(サブテーマ 1 と共通)
SMARCB1 が欠損したラブドイド腫瘍・類上皮肉腫の細胞では、SLC7A11 の転写が ARID1A 欠損がんと同様に強く減弱 していました。これにより、GSH 合成の必須基質である システイン の細胞内供給が慢性的に不足する状態にあります。
GCLC を律速酵素として標的化する設計
GSH 合成の経路で、システインとグルタミン酸を結合させて最初の中間体(γ-glutamylcysteine)を作る 律速酵素 が GCLC(Glutamate–Cysteine Ligase Catalytic subunit)です。私たちは、「システイン供給が既に綻んだ状態のがん細胞において、GCLC をさらに阻害すれば、GSH 合成の律速段階を直接遮断できる」 と仮説しました。
新規 GCLC 阻害剤 GCLCi1/GCLCi0 の創製(小野薬品工業との共同研究)
この仮説を検証するため、国立がん研究センターと小野薬品工業株式会社との共同研究 で 新規 GCLC 阻害剤 GCLCi1/GCLCi0 を創製しました。これらの化合物は、SMARCB1 欠損がん細胞において、細胞内 GSH を選択的に枯渇させることが確認されました。
作用機序
GCLC 阻害により枯渇した GSH は、GSH を補酵素とする酵素 GPX4(Glutathione Peroxidase 4)の活性低下を引き起こします。GPX4 は、細胞膜の 脂質過酸化物(脂質ヒドロペルオキシド)を還元して脂質を保護する役割を担っており、その活性が落ちると、細胞内の鉄(Fe²⁺)が触媒する Fenton 反応によって脂質過酸化が暴走 します。
この暴走的な脂質過酸化が、フェロトーシス(鉄依存性細胞死)と呼ばれる、アポトーシスとは異なる細胞死の形態を引き起こします。SWI/SNF 機能が保たれた正常細胞では、SLC7A11 発現が維持され GSH 合成にも余裕があるため、GCLC 阻害の影響は相対的に小さく、フェロトーシスは誘導されにくいと考えられます。これが、がん細胞選択性が得られる可能性 の理論的根拠となります。
検証
細胞株モデル(in vitro)
新規 GCLC 阻害剤 GCLCi1/GCLCi0 は、SMARCB1 欠損細胞株(悪性ラブドイド腫瘍細胞株、類上皮肉腫細胞株)に対して、SMARCB1 保持細胞株に比べて 高い選択的細胞死誘導活性 を示しました。
特筆すべき所見として、本研究の in vitro 評価系では、類上皮肉腫細胞において、GCLCi1/GCLCi0 は EZH2 阻害剤 Tazemetostat と比較して高い SMARCB1 欠損選択性を示しました。
フェロトーシス機構の検証
細胞死の形態が フェロトーシス(鉄依存性細胞死)であることを、以下の複数の独立した検証実験で確認しました。
- 脂質過酸化マーカー(C11-BODIPY、MDA 等)の上昇
- フェロトーシス阻害剤 Ferrostatin-1 による細胞死のレスキュー
- 細胞内 GSH の枯渇 の実測
- GPX4 活性低下 の確認
マウス xenograft モデル(in vivo)
悪性ラブドイド腫瘍のマウス異種移植モデル において、GCLCi1 単剤投与で有意な腫瘍増殖抑制 が確認されました。本実験条件下では、体重変化や主要臓器の組織病理学的所見において、顕著な毒性は観察されませんでした。
GCLC 阻害剤と GLS 阻害剤の合理的併用療法
GSH の合成には、システインに加えて グルタミン酸 も必須基質として必要です。SMARCB1 欠損がんにおける既存の「システイン供給の綻び」(SLC7A11 低下)に追い打ちをかける合理的な戦略として、私たちは グルタミン酸供給を絶つ GLS(グルタミナーゼ)阻害剤 Telaglenastat(CB-839) と、新規 GCLC 阻害剤 GCLCi1 を併用する代謝経路同時阻害療法を考案しました。
このアプローチでは、GSH 合成の 2 つの基質供給経路を同時に塞ぐ ため、in vitro のみならず in vivo マウス異種移植モデルにおいても、単剤投与を有意に上回る抗腫瘍効果 が確認されました。「なぜそう組み合わせると効くか」を、代謝マップ上で論理的に設計したうえで実証した点が、この併用療法の特徴です。
創薬・臨床への展望
本サブテーマは、SMARCB1 欠損希少がんに対する 新規治療標的化 の科学的基盤を提供するものです。想定される対象患者層は、悪性ラブドイド腫瘍の小児患者さん、類上皮肉腫の若年成人~成人患者さん となります。
創薬開発については、新規 GCLC 阻害剤 GCLCi1/GCLCi0 は、国立がん研究センターと小野薬品工業株式会社との共同研究 により創製されました(Takeuchi et al., Cancer Res, 2026)。本論文の謝辞・著者所属・本文中で同社との共同研究が公開されています。
また、GLS 阻害剤 Telaglenastat(CB-839) は、腎細胞がんなど複数のがん種で臨床試験が行われてきた薬剤であり、SWI/SNF 欠損がんへの応用可能性を検討しやすい候補と考えられます。新規 GCLC 阻害剤との合理的併用療法 は、研究室が代謝マップ上で論理的に設計した戦略として、今後の臨床開発の方向性を示しています。
研究室では、本概念を基盤として、SWI/SNF 欠損がん全般(→ 本ページ「サブテーマ 3」参照)への適応拡大、SWI/SNF 機能状態を反映する 層別化バイオマーカー の整備、既存療法との合理的併用療法の最適化、等の方向への展開を進めています。
関連論文
- Takeuchi M, Ishikawa Y, Okada T, Kozaki R, Ogiwara H*. A GCLC Inhibitor Enhances the Antitumor Efficacy of Glutathione Metabolic Pathway Inhibition in SMARCB1-Deficient Rhabdoid Tumors. Cancer Res. 2026;86(12):2990-3004. PubMed → SMARCB1 欠損希少がん(悪性ラブドイド腫瘍・類上皮肉腫)におけるグルタチオン代謝脆弱性の作用機序を解析。新規 GCLC 阻害剤 GCLCi1/GCLCi0 が、フェロトーシス(鉄依存性細胞死)を介した強い抗腫瘍効果を示すことを実証。小野薬品工業株式会社との共同研究成果。
関連プレスリリース
- 2026 年 3 月 17 日: SMARCB1 欠損希少がんの新たな治療標的を発見:グルタチオン代謝を標的とした GCLC 阻害剤のフェロトーシス誘導を介した新たな作用機序を解明 → プレスリリース
サブテーマ 3: SWI/SNF 欠損がん全般への普遍化 ── 共通病態に基づく対象がん横断的展開
研究要約
|
項目 |
内容 |
|---|---|
|
対象がん |
SWI/SNF クロマチンリモデリング複合体の構成因子欠損がん全般: ARID1A、SMARCA4、SMARCB1、PBRM1 欠損がん 等(卵巣明細胞がん、子宮体がん、胃がん、悪性ラブドイド腫瘍、類上皮肉腫、非小細胞肺がん、腎細胞がん 等を含む) |
|
遺伝子背景 |
SWI/SNF クロマチンリモデリング複合体の 構成因子の機能喪失(複数のサブユニットに横断的) |
|
標的・経路 |
サブテーマ 1 と共通: GSH 代謝経路/SLC7A11/GCLC/GPX4 |
|
作用機序 |
サブテーマ 1 と共通: SWI/SNF 機能喪失 → SLC7A11 転写低下 → システイン供給不足 → GSH 合成の綻び → GSH 阻害剤への高感受性(詳細はサブテーマ 1「作用機序」を参照) |
|
創薬上の意義 |
SLC7A11 低下を介する GSH 代謝脆弱性が、SWI/SNF 構成因子の違いを超えて共通する ことを実証。「単一遺伝子の変異」ではなく「SWI/SNF 複合体機能状態」を層別化概念として、複数のがん種に共通する治療標的化の枠組みとなる可能性を提示 |
|
検証段階 |
in vitro(4 つの SWI/SNF 因子欠損がん細胞株群での Eprenetapopt 感受性の系統的比較)/機構検証(SLC7A11 発現低下の SWI/SNF 因子横断的な確認) |
|
創薬段階 |
サブテーマ 1 と共通の研究ツール化合物 Eprenetapopt を使用 |
|
代表論文 |
Sasaki et al., Sci Rep, 2024;14(1):31321. PubMed |
このサブテーマの要点
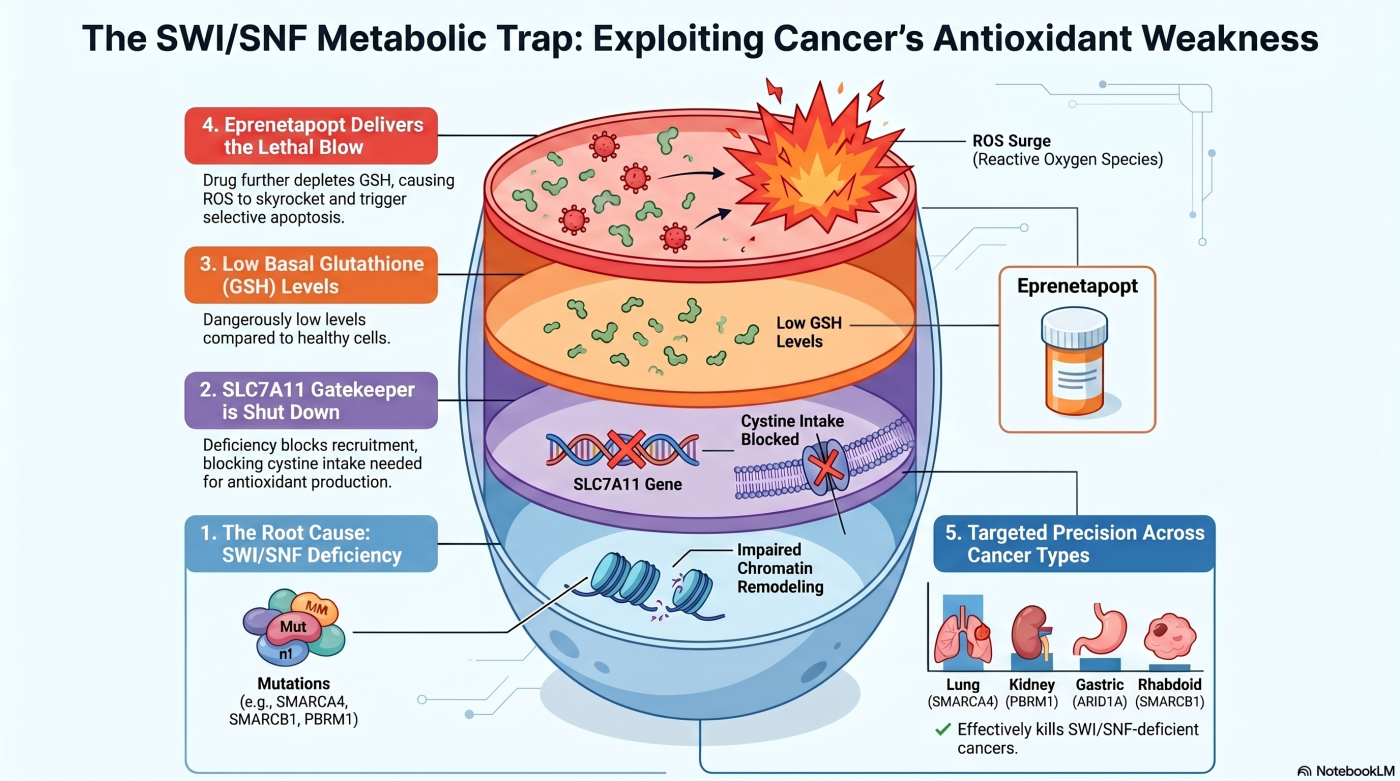
サブテーマ 1 で確立した「ARID1A 欠損 → SLC7A11 低下 → GSH 代謝脆弱性」という機序が、SWI/SNF クロマチンリモデリング複合体の構成因子の違いを超えて、共通する病態である ことを示しました(Sasaki et al., Sci Rep, 2024)。
ARID1A だけでなく、SMARCA4・SMARCB1・PBRM1 が欠損したがん細胞でも、SLC7A11 の発現低下 が共通して観察され、これら 4 つの SWI/SNF 因子のいずれかが欠損したがん細胞は、Eprenetapopt をはじめとする GSH 標的薬剤に対して、SWI/SNF 機能保持型細胞株と比較して高い感受性 を示しました。
本サブテーマは、SWI/SNF 複合体が 構成因子の違いを超えて、SLC7A11 転写を維持する共通の機能ハブとして働いている ことを示すとともに、「単一遺伝子の変異」ではなく「SWI/SNF 複合体の機能状態」を治療層別化の基準とする 新しい考え方を提示しています。
検証
私たちは、4 つの SWI/SNF 因子(ARID1A・SMARCA4・SMARCB1・PBRM1)のいずれかが欠損したがん細胞株群に対して、Eprenetapopt の感受性を系統的に比較 しました。その結果、いずれの SWI/SNF 因子の欠損型細胞株も、SWI/SNF 機能保持型細胞株と比較して Eprenetapopt に対して高い感受性 を示しました。
また、これら 4 つの SWI/SNF 因子欠損がん細胞株において、SLC7A11 mRNA 発現低下 が共通して観察され、SLC7A11 が SWI/SNF 因子横断的な機能ハブであることが確認されました。
なお、作用機序(SLC7A11 低下 → システイン供給不足 → GSH 合成の綻び → GSH 阻害剤への高感受性)はサブテーマ 1 と共通です。詳細は サブテーマ 1「作用機序」 をご参照ください。
創薬・臨床への展望
本サブテーマの結果は、「SWI/SNF 複合体の機能不全」を層別化概念として、複数のがん種に共通する治療標的化の枠組みとなる可能性 を示しています。想定される対象患者層は、ARID1A 欠損 OCCC・びまん性胃がん・子宮体がん、SMARCA4 欠損 非小細胞肺がん、SMARCB1 欠損 悪性ラブドイド腫瘍・類上皮肉腫、PBRM1 欠損 腎細胞がん 等、SWI/SNF 構成因子のいずれかが欠損したがんを持つ患者さん となります。
臨床での層別化(どの患者さんが本治療の恩恵を受けるか)にあたっては、単一遺伝子の変異だけでなく、SWI/SNF 複合体としての機能状態を示すバイオマーカー が重要になると考えています。研究室では、この層別化バイオマーカーの整備を、サブテーマ 1・2 と並行して進めています。
関連論文
- Sasaki M and Ogiwara H*. Efficacy of glutathione inhibitor eprenetapopt against the vulnerability of glutathione metabolism in SMARCA4-, SMARCB1- and PBRM1-deficient cancer cells. Sci Rep. 2024;14(1):31321. PubMed → サブテーマ 1(ARID1A 欠損がんでの発見)を、SWI/SNF 複合体欠損がん全般(ARID1A・SMARCA4・SMARCB1・PBRM1)に普遍化。SLC7A11 発現低下が共通の機構として働くことを実証。
キーターム
本ページ全体で使用する主要用語の解説です。
- グルタチオン代謝脆弱性: SWI/SNF クロマチンリモデリング複合体の構成因子欠損により生じる、GSH 合成経路の脆弱化を治療標的化する研究室の独自アプローチ
- グルタチオン(GSH): 細胞内で活性酸素種(ROS)と脂質過酸化物を中和する、最も主要な抗酸化分子。酸化ストレスに対する細胞の防御の中核を担う
- SLC7A11(シスチントランスポーター): 細胞外からシスチン(GSH の必須基質となる「システイン」の前駆体)を取り込む膜輸送体。SWI/SNF 複合体が転写を維持する標的遺伝子であり、本テーマの中核分子
- GCLC(Glutamate–Cysteine Ligase Catalytic subunit): グルタチオン合成の 律速酵素。システインとグルタミン酸を結合させて GSH 合成の最初の中間体(γ-glutamylcysteine)を作る
- GPX4(Glutathione Peroxidase 4): GSH を補酵素として、細胞膜の脂質過酸化物(脂質ヒドロペルオキシド)を還元する酵素。GSH 枯渇によって活性低下し、フェロトーシスを誘導する
- フェロトーシス(Ferroptosis): 細胞膜の 脂質過酸化 が暴走することで生じる、鉄依存性の細胞死 の一形態。アポトーシスとは異なる細胞死様式
- Eprenetapopt: GSH 枯渇・酸化ストレス誘導作用を持つ研究ツール化合物。本研究で ARID1A 欠損がんにおける GSH 代謝脆弱性の概念実証に使用
- 新規 GCLC 阻害剤 GCLCi1/GCLCi0: 国立がん研究センターと小野薬品工業株式会社との共同研究で創製された GCLC 阻害剤。SMARCB1 欠損希少がんに対するフェロトーシス誘導を介した抗腫瘍効果を示す
- GLS 阻害剤 Telaglenastat(CB-839): グルタミナーゼを標的とする低分子阻害剤。複数のがん種で臨床試験が行われてきた薬剤であり、本研究では新規 GCLC 阻害剤との合理的併用療法に用いられる
本ページの研究段階について
本ページで紹介する研究はいずれも、現時点では 基礎研究・前臨床研究段階の成果 です。GSH 代謝経路を標的とする治療薬は、現在臨床で使用可能な治療法ではありません。新規 GCLC 阻害剤 GCLCi1/GCLCi0 の実用化には、安全性・薬物動態評価、臨床試験など、複数の段階が必要となります。GLS 阻害剤 Telaglenastat(CB-839)は他のがん種で臨床試験が行われてきた薬剤ですが、本研究で扱う SWI/SNF 欠損がんに対する適応拡大は研究段階にあります。
関連ハイライト・ページ
- 従来型合成致死 ── 1 対 1 対応型 の合成致死戦略。研究室のパラログ合成致死研究の起点
- パラログ同時阻害法 ── 1 対 2 対応型 の合成致死戦略。SWI/SNF 欠損がんへのもう一つの主要アプローチ(CBP/p300 同時阻害)。本ページの代謝アプローチと並ぶ、研究室の主軸
- 次世代型合成致死 ── 2 対 n 対応型 への概念拡張(Dual SMARCA4/SMARCA2 欠損 × CHD3 阻害 等)
- データ駆動型探索 ── 公共データから新規標的を抽出する独自アプローチ。ARID1A 欠損 OCCC を共通の対象がん種とする並行研究系譜
- 既存薬の再定義 ── ARID1A 欠損がんに対するもう一つの代謝アプローチ。ピリミジン代謝の脆弱性を突く既存薬ゲムシタビンの選択的有効性
- 研究ハイライト(親) ── 6 つの研究テーマの俯瞰
- 研究プロジェクト ── 対象がん種別の研究プロジェクト(Project A-1: 悪性ラブドイド腫瘍/類上皮肉腫、Project A-2: 卵巣明細胞がん を含む)
- 論文業績 ── 全論文・総説・受賞歴
- がん治療学研究分野

