トップページ > 研究組織一覧 > 分野・独立ユニットグループ > がん治療学研究分野 > 研究成果の概要 > 従来型合成致死性
従来型合成致死性
メインテーマ概要
従来型合成致死とは
本ページでは、がんで失われた遺伝子の「もう一方のパラログ」を標的化することで、がん細胞だけに脆弱性を生じさせる 研究成果を紹介します。
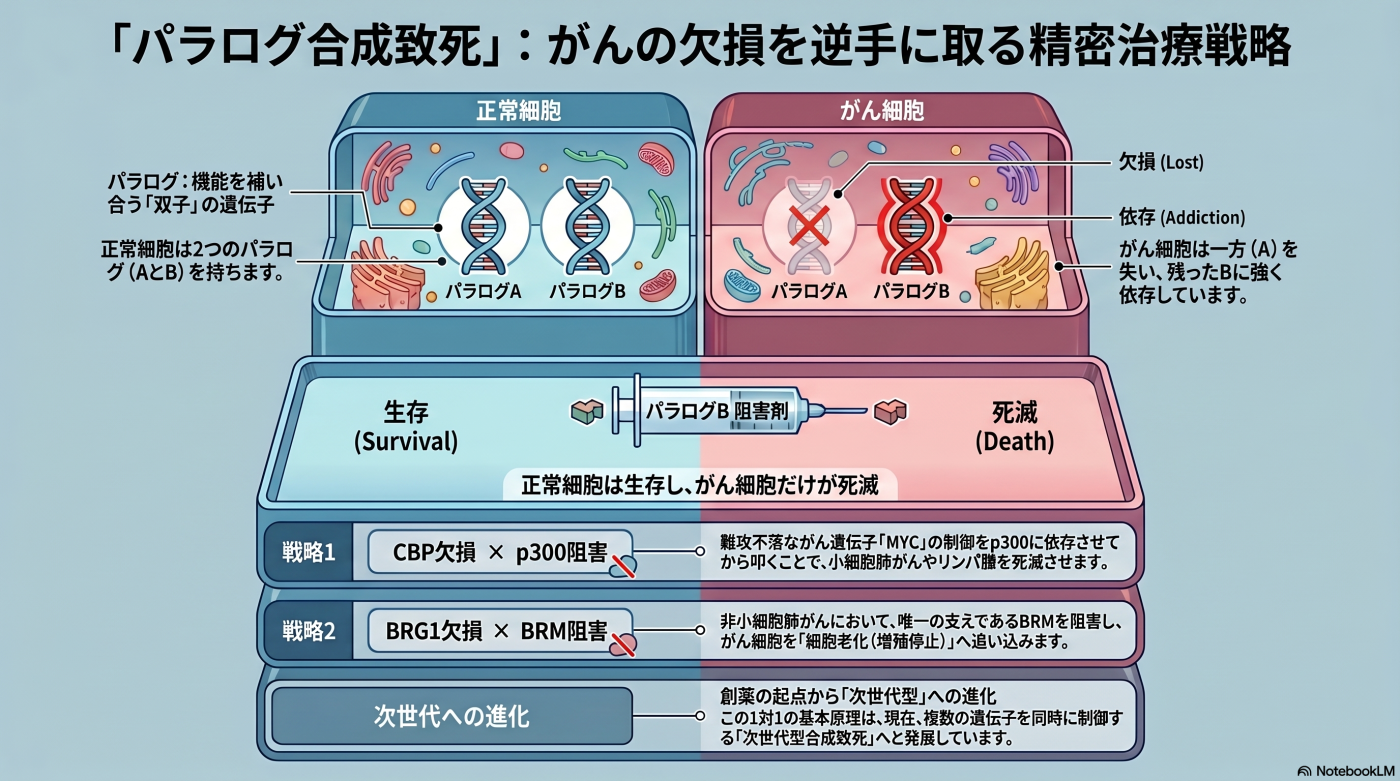
従来型合成致死 は、1 対 1 対応型 の合成致死戦略を指します。「がん細胞で欠損した 1 つの遺伝子 A」に対して、「別の 1 つの遺伝子 B」を阻害することで、正常細胞への影響を抑えながら、がん細胞を選択的に死滅させることを目指す戦略です。
本テーマで扱うのは、特に 「パラログ間の合成致死性」 に基づく従来型合成致死 ── すなわち、A と B がパラログ(互いに機能を補い合う類似遺伝子)の関係にある特殊ケース です。「がんで欠損したパラログ A」に対して、「もう一方のパラログ B」を阻害することで、補完機能を断ち切り、合成致死性を誘導します。
この概念は、その後の研究室の研究系譜において 次世代型合成致死(1 対 2 対応型のパラログ同時阻害法、および 2 対 n 対応型の次世代型合成致死)へと拡張されています。
研究室の研究系譜での位置づけ
本テーマは、研究室の 独自概念の起点 に位置する研究系譜です。2013 年 Cancer Res(BRG1/BRM)・2016 年 Cancer Discov(CBP/p300 addiction)で蓄積した「パラログ間の合成致死性」の理解と、SWI/SNF 関連変異がん細胞株パネル は、後続の主要な展開 ── パラログ同時阻害法、グルタチオン代謝脆弱性、データ駆動型探索、そして 次世代型合成致死 ── の共通基盤として、現在も研究室の中心に位置しています。
このページに掲載する研究
本ページでは、研究室の現在の研究系譜を形成した CREBBP/EP300 研究をサブテーマ1 に、これに先立ってパラログ合成致死性を実証した SMARCA4/SMARCA2 研究をサブテーマ2 に配置しています。
- サブテーマ 1: CREBBP 欠損がん × EP300 標的化(CBP/p300 パラログ)── Ogiwara et al., Cancer Discov, 2016。PI(荻原)筆頭著者論文 であり、研究室の研究系譜の起点 となった成果
- サブテーマ 2: SMARCA4 欠損非小細胞肺がん × SMARCA2 標的化(BRG1/BRM パラログ)── Oike et al., Cancer Res, 2013。パラログ合成致死研究系譜の最初の実証
サブテーマ 1: CREBBP 欠損がん × EP300 標的化 ── 研究室の研究系譜の起点
研究要約
|
項目 |
内容 |
|---|---|
|
対象がん |
CREBBP 変異がん: 小細胞肺がん、B 細胞性悪性リンパ腫(びまん性大細胞型 B 細胞リンパ腫、濾胞性リンパ腫 等)、膀胱がん 等 |
|
遺伝子背景 |
CREBBP(CBP)の機能喪失型変異 ── ヒストン H3K27 アセチル化酵素の欠損 |
|
標的・経路 |
EP300(p300) ── CREBBP のパラログ/MYC 転写維持経路(MYC プロモーターの H3K27 アセチル化を介する転写制御) |
|
作用機序 |
p300 阻害 → MYC プロモーターの H3K27ac 消失 → MYC 発現の維持破綻 → アポトーシス誘導 |
|
創薬上の意義 |
「パラログ間の合成致死性」をがん治療標的として実証 した成果。創薬が困難とされてきた MYC を間接的に制御する手段 として、p300 阻害を提示。本研究は、研究室の現在の研究系譜の起点となった成果 |
|
検証段階 |
in vitro(細胞株での選択的細胞死)/in vivo(マウス移植モデルでの腫瘍増殖抑制)/メカニズム解析(MYC 経路の制御機構解明) |
|
創薬段階 |
既知の p300/CBP 阻害剤として知られる研究ツール化合物 C646 を機能検証に用いた(Ogiwara et al., Cancer Discov 2016)/EP300 選択的阻害剤の創薬開発 が関連分野で進められており、その一部は公開文献で報告されている(J Med Chem 2023) |
|
代表論文 |
このサブテーマの要点
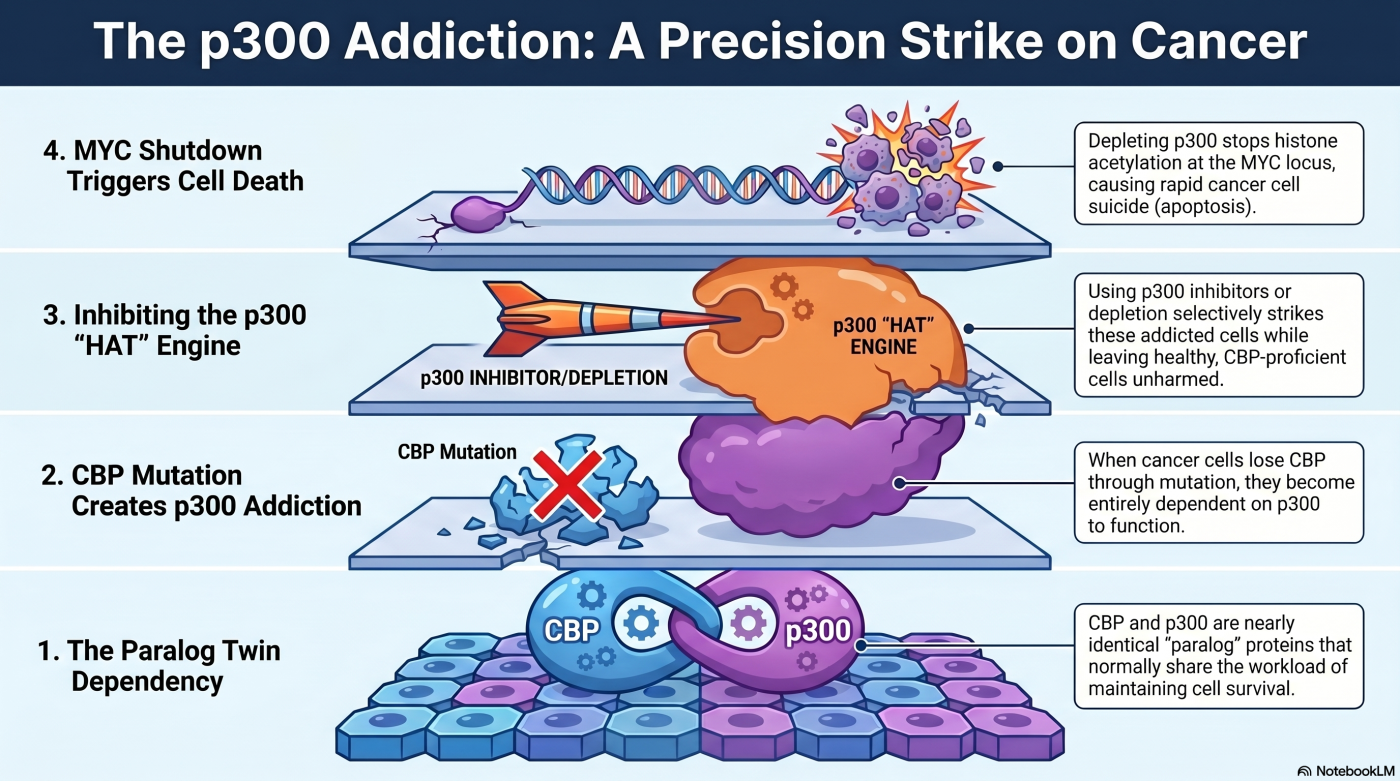
私たちは、CREBBP(CBP)が機能喪失している 小細胞肺がん・B 細胞性悪性リンパ腫 等の細胞が、生存のために CREBBP のパラログである EP300(p300)の機能に強く依存(addiction) していることを発見しました(Ogiwara et al., Cancer Discov, 2016)。
CBP と p300 は、ヒストン H3K27 をアセチル化する転写共役因子のパラログペアです。通常は両者が協調して、がん遺伝子 MYC の転写を維持しています。しかし、CBP が機能喪失すると、MYC の発現維持を p300 が単独で担う ようになり、結果として p300 への極端な依存性が生じます。p300 阻害は、創薬が困難とされてきた MYC を、間接的に制御する手段 として機能し、CBP 変異がんに対して選択的なアポトーシスを誘導します。
本研究は、PI(荻原)自身が筆頭著者として発表した、研究室の現在の研究系譜の起点となる成果 です。「パラログのもう片方を阻害すれば、がん抑制遺伝子の機能喪失すらも治療標的化できる」という発想と、それを実証する SWI/SNF 関連変異がん細胞株パネル は、この時に確立されました。
背景
本研究で取り組んだ CREBBP(CBP)変異がん は、有効な分子標的治療の選択肢が限られている、難治性の固形がん・血液がんです。
- 小細胞肺がん: 約 15% の症例で CREBBP/EP300 の体細胞変異
- B 細胞性悪性リンパ腫(びまん性大細胞型 B 細胞リンパ腫、濾胞性リンパ腫 等): CREBBP の機能喪失変異が頻発
- 膀胱がん ほか、複数のがん種でも CREBBP 変異が確認されている
CREBBP(CBP)は、EP300(p300)とともにヒストン H3K27 のアセチル化を担う転写共役因子 であり、エンハンサー活性や増殖関連遺伝子の発現制御に関与しています。その機能喪失はクロマチン状態の広範な変化と、MYC を含む増殖関連遺伝子群の発現異常を引き起こします。一方で、CREBBP 自体は機能喪失変異であり、従来の分子標的薬の概念では「打つべき標的がない」がんとして位置づけられてきました。
これらのがんは、既存化学療法・放射線療法に対する抵抗性症例が多く、既存の分子標的薬で直接標的化できるドライバー異常に乏しい ため、再発・転移例の予後は不良です。新たな治療選択肢の発見が長く望まれてきました。
発見
パラログという冗長性 ── 「失われた遺伝子」を治療標的化する発想
ヒトの遺伝子には、機能が重複する パラログ(類似遺伝子) が広く存在し、互いの機能を補い合っています。この 冗長性(redundancy) は、生体の頑健性を支える仕組みである一方、合成致死標的探索の壁ともなってきました。
私たちは、この冗長性を 逆手に取る ことを考えました。
正常細胞: パラログ A と B の両方が機能している。片方を阻害しても、もう一方が補完するため、細胞は生存する。
がん細胞(パラログ A 欠損型): A の機能を、残ったパラログ B に 強く依存(addiction) している。
治療戦略: B を阻害することで、がん細胞のバックアップを断ち切り、選択的な細胞死を誘導する ── これが 「パラログ間の合成致死性」に基づく従来型合成致死治療法 です。
CREBBP 欠損がんでの p300 依存と、MYC 経由の合成致死メカニズム
私たちは、CREBBP が機能喪失している小細胞肺がん・B 細胞性悪性リンパ腫細胞が、生存のために p300 の機能に強く依存 していることを発見し、その分子メカニズムを以下のように解明しました。
正常な細胞・非変異がん細胞: CBP と p300 が協調して、がん遺伝子 MYC の転写開始領域のヒストンをアセチル化し、その発現を維持している。
CBP 欠損がん細胞: CBP が失われた状態で、MYC の発現維持を p300 が単独で担っている。
p300 阻害時: MYC プロモーターの H3K27ac が消失し、MYC の発現が維持できなくなる → がん細胞は アポトーシス(細胞死) を起こして死滅する。
このメカニズムは、p300 阻害が MYC という「創薬が難しい(Undruggable)」標的を、間接的に制御する手段 として有効であることを示した点でも、独自性のある成果です。
作用機序
CBP 欠損がん細胞は、MYC 発現の維持を p300 に単独で依存している 状態にあります。p300 を阻害すると、
- MYC プロモーター領域の H3K27 アセチル化が消失
- MYC mRNA/タンパク質の発現が低下
- MYC 標的遺伝子群(細胞増殖関連遺伝子)の 転写出力が抑制
- がん細胞は アポトーシスを起こして死滅
という連続的な現象が誘導されます。これは、CBP 欠損がん細胞が 構造的に p300 へ依存(addiction) していることを示しており、CBP 機能が保たれた正常細胞や非変異がん細胞では、CBP と p300 の冗長性が維持されているため、p300 阻害の影響は相対的に小さくなります。これが、がん細胞選択性が得られる可能性 の理論的根拠となります。
検証
研究ツール化合物 C646 を用いた実証
私たちは、既知の p300/CBP 阻害剤として知られる研究ツール化合物 C646 を、本研究の機能検証に用いました(Ogiwara et al., Cancer Discov 2016)。C646 は研究用化合物であり、企業開発品とは独立した位置づけです。
細胞株モデル(in vitro)
C646 は、
- CBP 欠損 小細胞肺がん細胞株
- CBP 変異 B 細胞性悪性リンパ腫細胞株
に対して、用量依存的かつ選択的に高い殺細胞効果 を示しました。CBP を持つ非変異がん細胞・正常細胞には、同濃度域で生存への影響はほぼ認められませんでした。
マウス xenograft モデル(in vivo)
CBP 欠損腫瘍をマウスに移植したモデルに対し、p300 機能抑制で腫瘍増殖の有意な抑制と腫瘍縮小傾向 が観察されました。
メカニズム連動の検証
C646 投与後の腫瘍組織を解析したところ、
- MYC mRNA/タンパク質の発現低下
- MYC プロモーター領域の H3K27ac の減少
- アポトーシスマーカー(cleaved caspase-3 等)の上昇
が確認され、in vitro で同定した「p300 依存性 → MYC 発現破綻 → アポトーシス」経路が、in vivo 腫瘍モデルでも作動していることを支持 する結果が得られました。
創薬・臨床への展望
本研究は、CREBBP 変異がんに対する p300 を標的とした合成致死戦略 の科学的基盤を提供するものです。想定される対象患者層は、CREBBP 変異 小細胞肺がん患者さん、CREBBP 機能喪失 悪性リンパ腫患者さん(DLBCL、濾胞性リンパ腫の一部)、その他 CREBBP 変異が認められる固形がん患者さん(膀胱がん等)となります。
本研究で示したメカニズムに基づき、EP300 選択的阻害剤の創薬開発が関連分野で進められており、その一部は公開文献でも報告されています(J Med Chem 2023)。具体的な企業開発品の社内コード名・開発フェーズ・契約条件等については、知財および契約上の制約により本ページでは記載していません。
研究室では、本概念を基盤として、他の SWI/SNF 関連変異がん(ARID1A 欠損、PBRM1 欠損 等)への適応拡大、パラログ依存性を予測する 層別化バイオマーカー の整備、既存療法との合理的な併用療法の探索、次世代型合成致死(パラログ同時阻害法、1 対 2 対応型)への概念進化を含む方向への展開を進めています。
関連論文
- Ogiwara H, Sasaki M, Mitachi T, Oike T, Higuchi S, Tominaga Y, Kohno T*. Targeting p300 addiction in CBP-deficient cancers causes synthetic lethality via apoptotic cell death due to abrogation of MYC expression. Cancer Discov. 2016;6(4):430–445. PubMed → CREBBP(CBP)欠損がん(小細胞肺がん、悪性リンパ腫など)に対する EP300(p300)標的化と、MYC 抑制を介した合成致死メカニズムを解明。PI(荻原)自身が筆頭著者として発表した、研究室の現在の研究系譜の起点となる成果。
関連プレスリリース
- 2015 年 12 月 9 日: CBP 遺伝子変異がんに対する合成致死戦略の発見 → プレスリリース(上記 Ogiwara et al., Cancer Discov 2016 のオンライン公開時に対応する NCC 広報のプレスリリース)
サブテーマ 2: SMARCA4 欠損非小細胞肺がん × SMARCA2 標的化 ── パラログ合成致死研究系譜の最初の実証

研究要約
|
項目 |
内容 |
|---|---|
|
対象がん |
SMARCA4(BRG1)欠損 非小細胞肺がん(一部の症例で認められる) |
|
遺伝子背景 |
SMARCA4(BRG1)の機能喪失 ── SWI/SNF クロマチンリモデリング複合体の中核 ATPase 触媒サブユニットの欠損 |
|
標的・経路 |
SMARCA2(BRM) ── SMARCA4 のパラログ/SWI/SNF クロマチンリモデリング複合体の維持経路 |
|
作用機序 |
BRM 抑制 → SWI/SNF 複合体の 触媒機能の維持破綻 → 細胞老化(Senescence)誘導、増殖停止 |
|
創薬上の意義 |
「パラログ間の合成致死性」を実証した研究室の最初の研究。SWI/SNF 関連変異がん細胞株パネルの確立と、後続のパラログ合成致死研究系譜の出発点 |
|
検証段階 |
in vitro(細胞株での細胞老化誘導・増殖停止)/in vivo(マウス移植モデルでの腫瘍増殖抑制) |
|
創薬段階 |
SMARCA2 選択的阻害剤・分解誘導剤の創薬開発 が国際的に進行中 |
|
代表論文 |
このサブテーマの要点
私たちは、SMARCA4(BRG1)が機能喪失している 非小細胞肺がん 細胞において、SWI/SNF 複合体の機能維持を そのパラログである SMARCA2(BRM)が一手に担っている ことに着目しました。BRM の発現を抑制すると、BRG1 欠損肺がん細胞は 細胞老化(Senescence)に陥り、増殖が停止 します(Oike et al., Cancer Res, 2013)。
BRG1(SMARCA4)と BRM(SMARCA2)は、いずれも SWI/SNF クロマチンリモデリング複合体の ATPase 触媒サブユニット であり、構造・酵素活性ともに極めて酷似したパラログペアです。SWI/SNF 複合体には、BRG1 と BRM が排他的に組み込まれる ── 1 つの複合体には片方だけが入る構造になっており、互いに機能を補い合っています。BRG1 が欠損したがん細胞では、SWI/SNF 機能を BRM が単独で支えているため、BRM への極端な依存性が生じます。
本研究は、研究室の パラログ合成致死研究系譜の出発点となった実証研究 であり、後の CREBBP/EP300 研究(サブテーマ 1)、そして パラログ同時阻害法・次世代型合成致死 へとつながる研究系譜の起点となりました。
背景
本研究で取り組んだ SMARCA4(BRG1)欠損 非小細胞肺がん は、一部の症例で認められる難治性病型です。
- 非小細胞肺がん(NSCLC)の 一部の症例 で SMARCA4(BRG1)の機能喪失が認められる
- これらの症例の多くは、EGFR 変異・ALK 融合など、既存の分子標的薬の対象となる代表的ドライバー異常を持たない
- 喫煙関連の侵襲的腫瘍であることが多く、標準治療への反応性や予後には課題が残る症例が多い
SMARCA4 は、SWI/SNF クロマチンリモデリング複合体の ATPase 触媒サブユニット であり、SWI/SNF 機能の中核を担います。その機能喪失は、cBAF を含む複数の SWI/SNF サブタイプに広範な影響を及ぼします。一方で、SMARCA4 自体は機能喪失変異であり、従来の分子標的薬の概念では「打つべき標的がない」がんとして位置づけられてきました。
発見
着目したパラログペア:BRG1 と BRM
BRG1(SMARCA4)と BRM(SMARCA2) は、いずれも SWI/SNF クロマチンリモデリング複合体の ATPase 触媒サブユニット であり、構造・酵素活性ともに極めて酷似したパラログペアです。重要な特徴は、SWI/SNF 複合体には BRG1 と BRM が排他的に組み込まれる という点です ── 1 つの複合体には片方だけが入る構造になっており、互いに機能を補い合っています。
BRG1 欠損肺がんでの BRM 依存
私たちは、BRG1 が機能喪失している非小細胞肺がん細胞において、SWI/SNF 複合体の機能維持を BRM が一手に担っている ことに着目しました。BRM の発現を抑制すると、
- BRG1 欠損肺がん細胞は細胞老化(Senescence)に陥り、増殖が停止
- BRG1 を持つ正常細胞・非標的がん細胞では、BRM 抑制の影響は軽微
- マウス移植モデルでも、BRM 抑制により BRG1 欠損腫瘍の増殖が有意に抑制
されることを示しました。
これは、BRM の ATPase 活性を阻害する薬剤 が、BRG1 欠損肺がんに対する選択的治療薬となりうることを示した実証研究です。研究室のパラログ合成致死研究系譜の 出発点となる研究 として、後続のすべての研究の基盤となりました。
作用機序
BRG1 欠損がん細胞では、SWI/SNF クロマチンリモデリング機能を BRM が単独で支える 状態となります。BRM を阻害すると、
- SWI/SNF 複合体の ATPase 触媒機能が破綻
- クロマチンリモデリング能力が 広範に喪失
- 細胞周期関連遺伝子の発現異常を介して 細胞老化(Senescence)が誘導
- がん細胞の増殖が停止
という連続的な現象が誘導されます。これは、BRG1 欠損がん細胞が 構造的に BRM へ依存(addiction) していることを示しており、BRG1 機能が保たれた細胞では BRG1 と BRM の冗長性が維持されているため、BRM 抑制の影響は相対的に小さくなります。これが、がん細胞選択性が得られる可能性 の理論的根拠となります。
検証
細胞株モデル(in vitro)
BRG1 欠損肺がん細胞株での BRM 抑制により、
- 細胞老化マーカー(SA-β-gal、p21 等)の上昇
- 細胞増殖の停止
が確認されました。一方、BRG1 を持つ非変異肺がん細胞株・正常気道上皮細胞では、BRM 抑制の影響は軽微でした。
マウス xenograft モデル(in vivo)
マウス移植モデルでの BRG1 欠損腫瘍に対し、BRM 抑制で腫瘍増殖が有意に抑制 されました。
創薬・臨床への展望
本研究は、SMARCA4(BRG1)欠損 非小細胞肺がんに対する SMARCA2(BRM)を標的とした合成致死戦略 の科学的基盤を提供するものです。想定される対象患者層は、SMARCA4 欠損 非小細胞肺がん患者さんとなります。
本研究の発見に基づき、SMARCA2 選択的阻害剤・SMARCA2 分解誘導剤の創薬開発 が、国際的に進行中です。具体的な企業開発品の社内コード名・開発フェーズ・契約条件等については、知財および契約上の制約により本ページでは記載していません。
なお、SMARCA4 と SMARCA2 の 両方が同時に欠損した(dual loss) がんに対しては、SMARCA2 標的化戦略が適用できないため、別途異なる治療戦略が必要となります。研究室では、この dual loss 型のがんに対する新規合成致死標的として CHD3 を同定し、次世代型合成致死(2 対 1 対応型)として展開しています。
関連論文
- Oike T, Ogiwara H, Tominaga Y, Sasaki M, Itoh H, Nagayama Y, Yoshimura T, Itami T, Hirohashi Y, Saji H, Yoshida T, Kohno T*. A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1. Cancer Res. 2013;73(17):5508–5518. PubMed → SMARCA4(BRG1)欠損 非小細胞肺がんに対する SMARCA2(BRM)標的化を実証。研究室のパラログ合成致死研究系譜の 出発点となった実証研究。
キーターム
本ページ全体で使用する主要用語の解説です。
- 従来型合成致死(1 対 1 対応型合成致死性): 「がん細胞で欠損した 1 つの遺伝子」に対して「別の 1 つの遺伝子を阻害」する、2 因子間の合成致死戦略。本テーマで扱うのは、特に A と B がパラログ関係にある特殊ケース(パラログ間の合成致死性)
- パラログ(Paralog): 進化の過程で 1 つの遺伝子が複製され、機能が類似する 2 つのコピーとして並存している遺伝子ペアまたはグループ。互いに機能を補い合うため、片方の阻害だけでは治療効果が出にくいことが多い
- Addiction(依存): がん細胞が、本来は冗長な遺伝子に対して構造的に強く依存している状態。パラログ A が欠損した場合、もう一方のパラログ B への依存度が高まることがある
- SWI/SNF クロマチンリモデリング複合体: DNA を巻き付けたヒストンの構造を変えて、遺伝子のオン/オフを制御する分子装置。多くのヒトがんで構成因子の変異・欠損が認められる
- SMARCA4(BRG1)/SMARCA2(BRM): SWI/SNF 複合体の中核 ATPase 触媒サブユニットのパラログペア。互いに排他的に複合体へ組み込まれる
- CREBBP(CBP)/EP300(p300): ヒストン H3K27 をアセチル化する転写共役因子のパラログペア。構造・基質特異性ともに極めて酷似し、互いに機能を補い合う
- MYC: 細胞増殖を制御するがん遺伝子。直接的な薬理学的阻害が困難(Undruggable)とされてきたが、本研究では p300 阻害を介した 間接的な制御 が示された
- 細胞老化(Senescence): 細胞が増殖を不可逆的に停止する状態。BRM 抑制による BRG1 欠損がん細胞での誘導が、本研究の中核表現型
- 研究ツール化合物 C646: p300 を阻害する低分子化合物。Ogiwara et al., Cancer Discov 2016 等で公開された研究用化合物であり、企業開発品とは独立した位置づけ
本ページの研究段階について
本ページで紹介する研究はいずれも、現時点では 基礎研究・前臨床研究段階の成果 です。p300 阻害剤・SMARCA2 阻害剤等を標的とする治療薬は、現在臨床で使用可能な治療法ではありません。実用化には、阻害剤・分解誘導剤の開発、安全性・薬物動態評価、臨床試験など、複数の段階が必要となります。
関連ハイライト・ページ
- パラログ同時阻害法 ── 1 対 2 対応型(3 因子間の合成致死性) の戦略。本テーマの概念の 発展形
- 次世代型合成致死 ── 2 対 n 対応型 への概念拡張(Dual SMARCA4/SMARCA2 欠損 × CHD3 阻害 等)。本テーマの系譜のさらなる発展
- データ駆動型探索 ── 公共データから新規標的を抽出する独自アプローチ
- グルタチオン代謝脆弱性 ── 本研究で確立された SWI/SNF 関連変異がん細胞株パネルが基盤となった代謝研究
- 研究ハイライト(親) ── 6 つの研究テーマの俯瞰
- 研究プロジェクト ── 対象がん種別の研究プロジェクト
- 論文業績 ── 全論文・総説・受賞歴
最終更新日: 2026-05-16
- がん治療学研究分野

